Presence of a Mutation in PSEN1 Or PSEN2 Gene Is Associated with an Impaired Brain Endothelial Cell Phenotype in Vitro Snehal Raut, Ronak Patel and Abraham J
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Genetic Testing for Familial Alzheimer's Disease
Corporate Medical Policy Genetic Testing for Familial Alzheimer’s Disease AHS – M2038 File Name: genetic_testing_for_familial_alzheimers_disease Origination: 1/2019 Last CAP Review: 10/2020 Next CAP Review: 10/2021 Last Review: 10/2020 Description of Procedure or Service Alzheimer disease (AD) is a neurodegenerative disease defined by a gradual decline in memory, cognitive functions, gross atrophy of the brain, and an accumulation of extracellular amyloid plaques and intracellular neurofibrillary tangles (Karch, Cruchaga, & Goate, 2014). Familial Alzheimer disease (FAD) is a rare, inherited form of AD. FAD has a much earlier onset than other forms of Alzheimer disease with symptoms developing in individuals in their thirties or forties. Related Policies General Genetic Testing, Germline Disorders AHS – M2145 General Genetic Testing, Somatic Disorders AHS – M2146 ***Note: This Medical Policy is complex and technical. For questions concerning the technical language and/or specific clinical indications for its use, please consult your physician. Policy BCBSNC will provide coverage for genetic testing for familial Alzheimer disease when it is determined the medical criteria or reimbursement guidelines below are met. Benefits Application This medical policy relates only to the services or supplies described herein. Please refer to the Member's Benefit Booklet for availability of benefits. Member's benefits may vary according to benefit design; therefore member benefit language should be reviewed before applying the terms of this medical policy. -

Insurance and Advance Pay Test Requisition
Insurance and Advance Pay Test Requisition (2021) For Specimen Collection Service, Please Fax this Test Requisition to 1.610.271.6085 Client Services is available Monday through Friday from 8:30 AM to 9:00 PM EST at 1.800.394.4493, option 2 Patient Information Patient Name Patient ID# (if available) Date of Birth Sex designated at birth: 9 Male 9 Female Street address City, State, Zip Mobile phone #1 Other Phone #2 Patient email Language spoken if other than English Test and Specimen Information Consult test list for test code and name Test Code: Test Name: Test Code: Test Name: 9 Check if more than 2 tests are ordered. Additional tests should be checked off within the test list ICD-10 Codes (required for billing insurance): Clinical diagnosis: Age at Initial Presentation: Ancestral Background (check all that apply): 9 African 9 Asian: East 9 Asian: Southeast 9 Central/South American 9 Hispanic 9 Native American 9 Ashkenazi Jewish 9 Asian: Indian 9 Caribbean 9 European 9 Middle Eastern 9 Pacific Islander Other: Indications for genetic testing (please check one): 9 Diagnostic (symptomatic) 9 Predictive (asymptomatic) 9 Prenatal* 9 Carrier 9 Family testing/single site Relationship to Proband: If performed at Athena, provide relative’s accession # . If performed at another lab, a copy of the relative’s report is required. Please attach detailed medical records and family history information Specimen Type: Date sample obtained: __________ /__________ /__________ 9 Whole Blood 9 Serum 9 CSF 9 Muscle 9 CVS: Cultured 9 Amniotic Fluid: Cultured 9 Saliva (Not available for all tests) 9 DNA** - tissue source: Concentration ug/ml Was DNA extracted at a CLIA-certified laboratory or a laboratory meeting equivalent requirements (as determined by CAP and/or CMS)? 9 Yes 9 No 9 Other*: If not collected same day as shipped, how was sample stored? 9 Room temp 9 Refrigerated 9 Frozen (-20) 9 Frozen (-80) History of blood transfusion? 9 Yes 9 No Most recent transfusion: __________ /__________ /__________ *Please contact us at 1.800.394.4493, option 2 prior to sending specimens. -

Changes in Brain Transcripts Related to Alzheimer's Disease in a Model
Journal of Alzheimer’s Disease 30 (2012) 791–803 791 DOI 10.3233/JAD-2012-112183 IOS Press Changes in Brain Transcripts Related to Alzheimer’s Disease in a Model of HFE Hemochromatosis are not Consistent with Increased Alzheimer’s Disease Risk Daniel M. Johnstonea,b,c,e, Ross M. Grahamf,g,h, Debbie Trinderf,g, Carlos Riverosb,c,d, John K. Olynykf,g,i,j, Rodney J. Scotta,b,c, Pablo Moscatob,c,d and Elizabeth A. Milwarda,b,c,∗ aSchool of Biomedical Sciences and Pharmacy, The University of Newcastle, Callaghan, NSW, Australia bHunter Medical Research Institute, New Lambton Heights, NSW, Australia cPriority Research Centre for Bioinformatics, Biomarker Discovery and Information-Based Medicine, The University of Newcastle, Callaghan, NSW, Australia dSchool of Electrical Engineering and Computer Science, The University of Newcastle, Callaghan, NSW, Australia eBosch Institute and Discipline of Physiology, University of Sydney, NSW, Australia f School of Medicine and Pharmacology, University of Western Australia, Fremantle, WA, Australia gWestern Australian Institute for Medical Research, Perth, WA, Australia hSchool of Biomedical Sciences, Curtin University of Technology, Bentley, WA, Australia iCurtin Health Innovation Research Institute, Curtin University of Technology, Bentley, WA, Australia jDepartment of Gastroenterology, Fremantle Hospital, Fremantle, WA, Australia Accepted 5 March 2012 Abstract. Iron abnormalities are observed in the brains of Alzheimer’s disease (AD) patients, but it is unclear whether com- mon disorders of systemic iron overload such as hemochromatosis alter risks of AD. We used microarrays and real-time reverse transcription-PCR to investigate changes in the brain transcriptome of adult Hfe−/− mice, a model of hemochromatosis, relative to age- and gender-matched wildtype controls. -

Nanomedicine for Neurodegenerative Disorders: Focus on Alzheimer's
International Journal of Molecular Sciences Review Nanomedicine for Neurodegenerative Disorders: Focus on Alzheimer’s and Parkinson’s Diseases Keelan Jagaran and Moganavelli Singh * Nano-Gene and Drug Delivery Group, Discipline of Biochemistry, University of KwaZulu-Natal, Private Bag X54001, Durban 4000, South Africa; [email protected] * Correspondence: [email protected]; Tel.: +27-31-260-7170 Abstract: Neurodegenerative disorders involve the slow and gradual degeneration of axons and neurons in the central nervous system (CNS), resulting in abnormalities in cellular function and eventual cellular demise. Patients with these disorders succumb to the high medical costs and the disruption of their normal lives. Current therapeutics employed for treating these diseases are deemed palliative. Hence, a treatment strategy that targets the disease’s cause, not just the symptoms exhibited, is desired. The synergistic use of nanomedicine and gene therapy to effectively target the causative mutated gene/s in the CNS disease progression could provide the much-needed impetus in this battle against these diseases. This review focuses on Parkinson’s and Alzheimer’s diseases, the gene/s and proteins responsible for the damage and death of neurons, and the importance of nanomedicine as a potential treatment strategy. Multiple genes were identified in this regard, each presenting with various mutations. Hence, genome-wide sequencing is essential for specific treatment in patients. While a cure is yet to be achieved, genomic studies form the basis for creating a highly efficacious nanotherapeutic that can eradicate these dreaded diseases. Thus, nanomedicine can lead the way in helping millions of people worldwide to eventually lead a better life. -

The Untold Stories of the Speech Gene, the FOXP2 Cancer Gene
www.Genes&Cancer.com Genes & Cancer, Vol. 9 (1-2), January 2018 The untold stories of the speech gene, the FOXP2 cancer gene Maria Jesus Herrero1,* and Yorick Gitton2,* 1 Center for Neuroscience Research, Children’s National Medical Center, NW, Washington, DC, USA 2 Sorbonne University, INSERM, CNRS, Vision Institute Research Center, Paris, France * Both authors contributed equally to this work Correspondence to: Yorick Gitton, email: [email protected] Keywords: FOXP2 factor, oncogene, cancer, prognosis, language Received: March 01, 2018 Accepted: April 02, 2018 Published: April 18, 2018 Copyright: Herrero and Gitton et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License 3.0 (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT FOXP2 encodes a transcription factor involved in speech and language acquisition. Growing evidence now suggests that dysregulated FOXP2 activity may also be instrumental in human oncogenesis, along the lines of other cardinal developmental transcription factors such as DLX5 and DLX6 [1–4]. Several FOXP family members are directly involved during cancer initiation, maintenance and progression in the adult [5–8]. This may comprise either a pro- oncogenic activity or a deficient tumor-suppressor role, depending upon cell types and associated signaling pathways. While FOXP2 is expressed in numerous cell types, its expression has been found to be down-regulated in breast cancer [9], hepatocellular carcinoma [8] and gastric cancer biopsies [10]. Conversely, overexpressed FOXP2 has been reported in multiple myelomas, MGUS (Monoclonal Gammopathy of Undetermined Significance), several subtypes of lymphomas [5,11], as well as in neuroblastomas [12] and ERG fusion-negative prostate cancers [13]. -

Engineered Type 1 Regulatory T Cells Designed for Clinical Use Kill Primary
ARTICLE Acute Myeloid Leukemia Engineered type 1 regulatory T cells designed Ferrata Storti Foundation for clinical use kill primary pediatric acute myeloid leukemia cells Brandon Cieniewicz,1* Molly Javier Uyeda,1,2* Ping (Pauline) Chen,1 Ece Canan Sayitoglu,1 Jeffrey Mao-Hwa Liu,1 Grazia Andolfi,3 Katharine Greenthal,1 Alice Bertaina,1,4 Silvia Gregori,3 Rosa Bacchetta,1,4 Norman James Lacayo,1 Alma-Martina Cepika1,4# and Maria Grazia Roncarolo1,2,4# Haematologica 2021 Volume 106(10):2588-2597 1Department of Pediatrics, Division of Stem Cell Transplantation and Regenerative Medicine, Stanford School of Medicine, Stanford, CA, USA; 2Stanford Institute for Stem Cell Biology and Regenerative Medicine, Stanford School of Medicine, Stanford, CA, USA; 3San Raffaele Telethon Institute for Gene Therapy, Milan, Italy and 4Center for Definitive and Curative Medicine, Stanford School of Medicine, Stanford, CA, USA *BC and MJU contributed equally as co-first authors #AMC and MGR contributed equally as co-senior authors ABSTRACT ype 1 regulatory (Tr1) T cells induced by enforced expression of interleukin-10 (LV-10) are being developed as a novel treatment for Tchemotherapy-resistant myeloid leukemias. In vivo, LV-10 cells do not cause graft-versus-host disease while mediating graft-versus-leukemia effect against adult acute myeloid leukemia (AML). Since pediatric AML (pAML) and adult AML are different on a genetic and epigenetic level, we investigate herein whether LV-10 cells also efficiently kill pAML cells. We show that the majority of primary pAML are killed by LV-10 cells, with different levels of sensitivity to killing. Transcriptionally, pAML sensitive to LV-10 killing expressed a myeloid maturation signature. -

Genetic Testing for Neurologic Disorders MP9497
Coverage of any medical intervention discussed in a Dean Health Plan medical policy is subject to the limitations and exclusions outlined in the member's benefit certificate or policy and to applicable state and/or federal laws. Genetic Testing for Neurologic Disorders MP9497 Covered Service: Yes Prior Authorization Required: Yes Additional Genetic testing is covered for a Dean Health Plan member if Information: the test results provide a direct medical benefit or guides reproductive decision-making for the Dean Health Plan member. See Genetic Testing MP9012 for additional information. Pre- and post-test genetic counseling is required for any individual undergoing genetic testing for neurological disorders. Pre and post-genetic counseling is not required for ASO members. Please reference the ASO Summary Plan Document (SPD). A first-degree relative is defined as an individual’s parents, full siblings, and children. A second-degree relative is defined as an individual’s grandparents, grandchildren, aunts, uncles, nephews, nieces and half-siblings. *Axonal neuropathy indicates etiology is related to diabetes, toxic medications, or thyroid disease. Reproductive carrier screening (prenatal testing) does not require prior authorization and is addressed per MP9477 Medicare Policy: Prior authorization is dependent on the member’s Medicare coverage. Prior authorization is not required for Medicare Cost products (Dean Care Gold) and Medicare Supplement (Select) when this service is provided by participating providers. Prior authorization is required if a member has Medicare primary and Dean Health Plan secondary coverage. This policy is not applicable to our Medicare Replacement products. BadgerCare Plus Dean Health Plan covers when BadgerCare Plus also covers Policy: the benefit. -

Common Variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33
LETTERS Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease We sought to identify new susceptibility loci for Alzheimer’s (P = 3 × 10−3). A combined analysis of the GERAD1 and EADI1 data disease through a staged association study (GERAD+) yielded highly significant support for all three loci (CLU meta P = and by testing suggestive loci reported by the Alzheimer’s 6.7 × 10−16; PICALM meta P = 6.3 × 10−9; and CR1 meta P = 3.2 × Disease Genetic Consortium (ADGC) in a companion 10−12). The associations in CLU, PICALM and CRI have since been paper. We undertook a combined analysis of four genome- replicated in several independent datasets5–8, have shown trends in wide association datasets (stage 1) and identified ten newly another dataset9 and have shown relationships with the neurodegen- associated variants with P ≤ 1 × 10−5. We tested these variants erative processes underlying disease10. In addition, members of this for association in an independent sample (stage 2). Three SNPs consortium have since reported genome-wide significant association at two loci replicated and showed evidence for association in for BIN1 (P = 1.6 × 10−11) and support for EPHA1 (encoding ephrin a further sample (stage 3). Meta-analyses of all data provided receptor A1) (P = 1.7 × 10−6)11. compelling evidence that ABCA7 (rs3764650, meta P = 4.5 × This study sought to identify new common susceptibility variants 10−17; including ADGC data, meta P = 5.0 × 10−21) and the for Alzheimer’s disease by first undertaking a three-stage associa- All rights reserved. -

Genes and Pathways Associated with Pregnancy Loss in Dairy Cattle Anil Sigdel1, Rafael S
www.nature.com/scientificreports OPEN Genes and pathways associated with pregnancy loss in dairy cattle Anil Sigdel1, Rafael S. Bisinotto2 & Francisco Peñagaricano1* Pregnancy loss directly impairs reproductive performance in dairy cattle. Here, we investigated genetic factors associated with pregnancy loss following detection of a viable embryo around 42 days of gestation. The objectives of this study were to perform whole-genome scans and subsequent gene-set analyses for identifying candidate genes, functional gene-sets and gene signaling pathways implicated in pregnancy loss in US Holstein cows. Data consisted of about 58,000 pregnancy/ abortion records distributed over nulliparous, primiparous, and multiparous cows. Threshold models were used to assess the binary response of pregnancy loss. Whole‐genome scans identifed at least seven genomic regions on BTA2, BTA10, BTA14, BTA16, BTA21, BTA24 and BTA29 associated with pregnancy loss in heifers and lactating cows. These regions harbor several candidate genes that are directly implicated in pregnancy maintenance and fetal growth, such as CHST14, IGF1R, IGF2, PSEN2, SLC2A5 and WNT4. Moreover, the enrichment analysis revealed at least seven signifcantly enriched processes, containing genes associated with pregnancy loss, including calcium signaling, cell–cell attachment, cellular proliferation, fetal development, immunity, membrane permeability, and steroid metabolism. Additionally, the pathway analysis revealed a number of signifcant gene signaling pathways that regulate placental development and fetal growth, including Wnt, Hedgehog, Notch, MAPK, Hippo, mTOR and TGFβ pathways. Overall, our fndings contribute to a better understanding of the genetic and biological basis of pregnancy loss in dairy cattle and points out novel strategies for improving pregnancy maintenance via marker‐assisted breeding. -
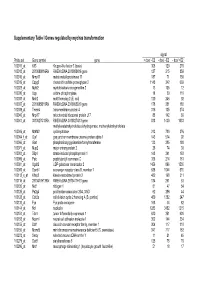
Supplementary Table I Genes Regulated by Myc/Ras Transformation
Supplementary Table I Genes regulated by myc/ras transformation signal Probe set Gene symbol gene + dox/ - E2 - dox/ - E2 - dox/ +E2 100011_at Klf3 Kruppel-like factor 3 (basic) 308 120 270 100013_at 2010008K16Rik RIKEN cDNA 2010008K16 gene 127 215 859 100016_at Mmp11 matrix metalloproteinase 11 187 71 150 100019_at Cspg2 chondroitin sulfate proteoglycan 2 1148 342 669 100023_at Mybl2 myeloblastosis oncogene-like 2 13 105 12 100030_at Upp uridine phosphorylase 18 39 110 100033_at Msh2 mutS homolog 2 (E. coli) 120 246 92 100037_at 2310005B10Rik RIKEN cDNA 2310005B10 gene 178 351 188 100039_at Tmem4 transmembrane protein 4 316 135 274 100040_at Mrpl17 mitochondrial ribosomal protein L17 88 142 89 100041_at 3010027G13Rik RIKEN cDNA 3010027G13 gene 818 1430 1033 methylenetetrahydrofolate dehydrogenase, methenyltetrahydrofolate 100046_at Mthfd2 cyclohydrolase 213 720 376 100064_f_at Gja1 gap junction membrane channel protein alpha 1 143 574 92 100066_at Gart phosphoribosylglycinamide formyltransferase 133 385 180 100071_at Mup2 major urinary protein 2 26 74 30 100081_at Stip1 stress-induced phosphoprotein 1 148 341 163 100089_at Ppic peptidylprolyl isomerase C 358 214 181 100091_at Ugalt2 UDP-galactose translocator 2 1456 896 1530 100095_at Scarb1 scavenger receptor class B, member 1 638 1044 570 100113_s_at Kifap3 kinesin-associated protein 3 482 168 311 100116_at 2810417H13Rik RIKEN cDNA 2810417H13 gene 134 261 53 100120_at Nid1 nidogen 1 81 47 54 100125_at Pa2g4 proliferation-associated 2G4, 38kD 62 286 44 100128_at Cdc2a cell division cycle 2 homolog A (S. pombe) 459 1382 247 100133_at Fyn Fyn proto-oncogene 168 44 82 100144_at Ncl nucleolin 1283 3452 1215 100151_at Tde1 tumor differentially expressed 1 620 351 620 100153_at Ncam1 neural cell adhesion molecule 1 302 144 234 100155_at Ddr1 discoidin domain receptor family, member 1 304 117 310 100156_at Mcmd5 mini chromosome maintenance deficient 5 (S. -

Genetics of Dementia Genetic Discovery Informs Our Understanding of Disease Mechanisms and Potential Drug Targets
DEMENTIA CARE APPROACHES Genetics of Dementia Genetic discovery informs our understanding of disease mechanisms and potential drug targets. An Interview With Rudolph E. Tanzi, PhD What Should Clinical Neurologists ple, if someone finds out they are a homozygote, they are also Know About ApoE4? finding out there is a 50% chance that their children will have Many clinicians may know that having 1 copy at least 1 ApoE4 allele and this can affect important life deci- of the apolipoprotein E e4 (ApoE4) allele (het- sions. Thus, the recommendation for genetic counseling. There erozygous) is the most common risk factor for are commercially available tests, and individuals can do as they late-onset Alzheimer disease (AD) with average wish, but we see no clinical reason for it at this time. age 70 at onset. Clinicians may not realize, however, that peo- ple who have 2 copies of ApoE4 (homozygotes) can have early What Are We Learning From Familial AD? onset under age 60 that appears similar to familial AD. People The genes that cause familial AD, including amyloid pre- with 1 copy of ApoE4 have a 3- to 4-times increased risk of AD; cursor protein (APP), which I discovered during my doctoral those with 2 copies have a 12- to 14-times higher risk of AD.1 research,3 and presenilin 1 and 2 (PSEN1/2), which Peter Importantly, however, having an ApoE4 allele does not guar- St George Hyslop, Gerard Schellenberg, and I found,3 have antee AD will develop because the gene is not fully penetrant. approximately 300 identified mutations.