Gene Copy Number Variation Spanning 60 Million Years of Human and Primate Evolution
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Recombinant Human Carbonic Anhydrase XIII Protein Catalog Number: ATGP3059
Recombinant human Carbonic Anhydrase XIII protein Catalog Number: ATGP3059 PRODUCT INPORMATION Expression system E.coli Domain 1-262aa UniProt No. Q8N1Q1 NCBI Accession No. NP_940986.1 Alternative Names Carbonic anhydrase 13, CAXIII PRODUCT SPECIFICATION Molecular Weight 31.8 kDa (285aa) confirmed by MALDI-TOF Concentration 0.5mg/ml (determined by Bradford assay) Formulation Liquid in. Phosphate-Buffered Saline (pH 7.4) containing 10% glycerol, 1mM DTT Purity > 90% by SDS-PAGE Biological Activity Specific activity is > 2,500pmol/min/ug, and is defined as the amount of enzyme that hydrolyze 1.0pmole of 4- nitrophenyl acetate to 4-nitrophenol per minute at pH 7.5 at 37C. Tag His-Tag Application SDS-PAGE, Enzyme Activity Storage Condition Can be stored at +2C to +8C for 1 week. For long term storage, aliquot and store at -20C to -80C. Avoid repeated freezing and thawing cycles. BACKGROUND Description CA13 also known as carbonic anhydrase 13 belongs to the alpha-carbonic anhydrase family. The carbonic anhydrase from a family of enzymes that catalyze the rapid interconversion of carbon dioxide and water to bicarbonate and protons, a reversible reaction that occurs relatively slowly in the absence of catalyst. The active 1 Recombinant human Carbonic Anhydrase XIII protein Catalog Number: ATGP3059 site of most carbonic anhydrases contains a zinc ion; they are claasified as metalloenzymes. There are at least five distinct CA families (alpha, beta, gamma, delta, and epsilon). These families have no significant amino acid sequence similarity and in most cases are thought to be an example of convergent evolution. The alpha-CAs are found in humans. -

Table 2. Significant
Table 2. Significant (Q < 0.05 and |d | > 0.5) transcripts from the meta-analysis Gene Chr Mb Gene Name Affy ProbeSet cDNA_IDs d HAP/LAP d HAP/LAP d d IS Average d Ztest P values Q-value Symbol ID (study #5) 1 2 STS B2m 2 122 beta-2 microglobulin 1452428_a_at AI848245 1.75334941 4 3.2 4 3.2316485 1.07398E-09 5.69E-08 Man2b1 8 84.4 mannosidase 2, alpha B1 1416340_a_at H4049B01 3.75722111 3.87309653 2.1 1.6 2.84852656 5.32443E-07 1.58E-05 1110032A03Rik 9 50.9 RIKEN cDNA 1110032A03 gene 1417211_a_at H4035E05 4 1.66015788 4 1.7 2.82772795 2.94266E-05 0.000527 NA 9 48.5 --- 1456111_at 3.43701477 1.85785922 4 2 2.8237185 9.97969E-08 3.48E-06 Scn4b 9 45.3 Sodium channel, type IV, beta 1434008_at AI844796 3.79536664 1.63774235 3.3 2.3 2.75319499 1.48057E-08 6.21E-07 polypeptide Gadd45gip1 8 84.1 RIKEN cDNA 2310040G17 gene 1417619_at 4 3.38875643 1.4 2 2.69163229 8.84279E-06 0.0001904 BC056474 15 12.1 Mus musculus cDNA clone 1424117_at H3030A06 3.95752801 2.42838452 1.9 2.2 2.62132809 1.3344E-08 5.66E-07 MGC:67360 IMAGE:6823629, complete cds NA 4 153 guanine nucleotide binding protein, 1454696_at -3.46081884 -4 -1.3 -1.6 -2.6026947 8.58458E-05 0.0012617 beta 1 Gnb1 4 153 guanine nucleotide binding protein, 1417432_a_at H3094D02 -3.13334396 -4 -1.6 -1.7 -2.5946297 1.04542E-05 0.0002202 beta 1 Gadd45gip1 8 84.1 RAD23a homolog (S. -

Steroid-Dependent Regulation of the Oviduct: a Cross-Species Transcriptomal Analysis
University of Kentucky UKnowledge Theses and Dissertations--Animal and Food Sciences Animal and Food Sciences 2015 Steroid-dependent regulation of the oviduct: A cross-species transcriptomal analysis Katheryn L. Cerny University of Kentucky, [email protected] Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Cerny, Katheryn L., "Steroid-dependent regulation of the oviduct: A cross-species transcriptomal analysis" (2015). Theses and Dissertations--Animal and Food Sciences. 49. https://uknowledge.uky.edu/animalsci_etds/49 This Doctoral Dissertation is brought to you for free and open access by the Animal and Food Sciences at UKnowledge. It has been accepted for inclusion in Theses and Dissertations--Animal and Food Sciences by an authorized administrator of UKnowledge. For more information, please contact [email protected]. STUDENT AGREEMENT: I represent that my thesis or dissertation and abstract are my original work. Proper attribution has been given to all outside sources. I understand that I am solely responsible for obtaining any needed copyright permissions. I have obtained needed written permission statement(s) from the owner(s) of each third-party copyrighted matter to be included in my work, allowing electronic distribution (if such use is not permitted by the fair use doctrine) which will be submitted to UKnowledge as Additional File. I hereby grant to The University of Kentucky and its agents the irrevocable, non-exclusive, and royalty-free license to archive and make accessible my work in whole or in part in all forms of media, now or hereafter known. -

(12) Patent Application Publication (10) Pub. No.: US 2003/0082511 A1 Brown Et Al
US 20030082511A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2003/0082511 A1 Brown et al. (43) Pub. Date: May 1, 2003 (54) IDENTIFICATION OF MODULATORY Publication Classification MOLECULES USING INDUCIBLE PROMOTERS (51) Int. Cl." ............................... C12O 1/00; C12O 1/68 (52) U.S. Cl. ..................................................... 435/4; 435/6 (76) Inventors: Steven J. Brown, San Diego, CA (US); Damien J. Dunnington, San Diego, CA (US); Imran Clark, San Diego, CA (57) ABSTRACT (US) Correspondence Address: Methods for identifying an ion channel modulator, a target David B. Waller & Associates membrane receptor modulator molecule, and other modula 5677 Oberlin Drive tory molecules are disclosed, as well as cells and vectors for Suit 214 use in those methods. A polynucleotide encoding target is San Diego, CA 92121 (US) provided in a cell under control of an inducible promoter, and candidate modulatory molecules are contacted with the (21) Appl. No.: 09/965,201 cell after induction of the promoter to ascertain whether a change in a measurable physiological parameter occurs as a (22) Filed: Sep. 25, 2001 result of the candidate modulatory molecule. Patent Application Publication May 1, 2003 Sheet 1 of 8 US 2003/0082511 A1 KCNC1 cDNA F.G. 1 Patent Application Publication May 1, 2003 Sheet 2 of 8 US 2003/0082511 A1 49 - -9 G C EH H EH N t R M h so as se W M M MP N FIG.2 Patent Application Publication May 1, 2003 Sheet 3 of 8 US 2003/0082511 A1 FG. 3 Patent Application Publication May 1, 2003 Sheet 4 of 8 US 2003/0082511 A1 KCNC1 ITREXCHO KC 150 mM KC 2000000 so 100 mM induced Uninduced Steady state O 100 200 300 400 500 600 700 Time (seconds) FIG. -

And Primate Evolution Gene Copy Number Variation Spanning 60
Downloaded from genome.cshlp.org on January 27, 2009 - Published by Cold Spring Harbor Laboratory Press Gene copy number variation spanning 60 million years of human and primate evolution Laura Dumas, Young H. Kim, Anis Karimpour-Fard, et al. Genome Res. 2007 17: 1266-1277 originally published online July 31, 2007 Access the most recent version at doi:10.1101/gr.6557307 Supplemental http://genome.cshlp.org/content/suppl/2007/07/26/gr.6557307.DC1.html Material References This article cites 51 articles, 20 of which can be accessed free at: http://genome.cshlp.org/content/17/9/1266.full.html#ref-list-1 Article cited in: http://genome.cshlp.org/content/17/9/1266.full.html#related-urls Email alerting Receive free email alerts when new articles cite this article - sign up in the box at the service top right corner of the article or click here To subscribe to Genome Research go to: http://genome.cshlp.org/subscriptions Copyright © 2007, Cold Spring Harbor Laboratory Press Downloaded from genome.cshlp.org on January 27, 2009 - Published by Cold Spring Harbor Laboratory Press Article Gene copy number variation spanning 60 million years of human and primate evolution Laura Dumas,1 Young H. Kim,2 Anis Karimpour-Fard,3 Michael Cox,1,4,5 Janet Hopkins,1,4,5 Jonathan R. Pollack,2 and James M. Sikela1,4,5,6 1Human Medical Genetics Program, University of Colorado at Denver and Health Sciences Center, Aurora, Colorado 80045, USA; 2Department of Pathology, Stanford University, Stanford, California 94305, USA; 3Department of Preventative Medicine and Biometrics, -

Identifying Nootropic Drug Targets Via Large-Scale Cognitive GWAS and Transcriptomics
bioRxiv preprint doi: https://doi.org/10.1101/2020.02.06.934752; this version posted February 6, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Title: Identifying Nootropic Drug Targets via Large-Scale Cognitive GWAS and Transcriptomics Max Lam1, 2, 3,4, Chia-Yen, Chen3,5,6, Xia Yan7,8, W. David Hill9, 10, Joey W. Trampush11, Jin Yu1, Emma Knowles12,13,14, Gail Davies9, 10, Eli Stahl15, 16, Laura Huckins15, 16, David C. Liewald10, Srdjan Djurovic17, 18, Ingrid Melle18, 19, Andrea Christoforou20, Ivar Reinvang21, Pamela DeRosse1, 22, 23, Astri J. Lundervold24, Vidar M. Steen18, 20, Thomas Espeseth19, 21, Katri Räikkönen25, Elisabeth Widen26, Aarno Palotie26, 27, 28, Johan G. Eriksson29, 30, 31, Ina Giegling32, Bettina Konte32, Annette M. Hartmann32, Panos Roussos15, 16, 33, Stella Giakoumaki34, Katherine E. Burdick15, 33, 35, Antony Payton36, William Ollier37, 38, Ornit Chiba- Falek39, Deborah K. Koltai39, 40 , Anna C. Need41, Elizabeth T. Cirulli42, Aristotle N. Voineskos43, Nikos C. Stefanis44, 45, 46, Dimitrios Avramopoulos47, 48, Alex Hatzimanolis44, 45, 46, Nikolaos Smyrnis44, 45, Robert M. Bilder49, Nelson A. Freimer49, Tyrone D. Cannon50, 51, Edythe London49, Russell A. Poldrack52, Fred W. Sabb53, Eliza Congdon49, Emily Drabant Conley54, Matthew A. Scult55, Dwight Dickinson56, Richard E. Straub57, Gary Donohoe58, Derek Morris58, Aiden Corvin59, Michael Gill59, Ahmad R. Hariri55, Daniel R. Weinberger57, Neil Pendleton60, Panos Bitsios61, Dan Rujescu32, Jari Lahti25, 62, Stephanie Le Hellard18, 20, Matthew C. -

S1 of S77 Supplementary Materials: Discovery of a New Class of Cathepsin Kinhibitors in Rhizoma Drynariaeas Potential Candidates for the Treatment of Osteoporosis
Int. J. Mol. Sci.2016, 17, 2116; doi:10.3390/ijms17122116 S1 of S77 Supplementary Materials: Discovery of a New Class of Cathepsin KInhibitors in Rhizoma Drynariaeas Potential Candidates for the Treatment of Osteoporosis Zuo-Cheng Qiu, Xiao-Li Dong, Yi Dai, Gao-Keng Xiao, Xin-Luan Wang, Ka-Chun Wong, Man-Sau Wong and Xin-Sheng Yao Table S1. Compounds identified from Drynariae rhizome (DR). No. Compound Name Chemical Structure 1 Naringin 5,7,3′,5′-Tetrahydroxy-flavanone 2 7-O-neohesperidoside 3 Narigenin-7-O-β-D-glucoside 5,7,3′,5′-Tetrahydroxy-flavanone 4 7-O-β-D-glucopyranoside 5 Naringenin 6 5,7,3′,5′-Tetrahydroxyflavanone 7 Kushennol F 8 Sophoraflavanone G 9 Kurarinone Int. J. Mol. Sci.2016, 17, 2116; doi:10.3390/ijms17122116 S2 of S77 Table S1. Cont. No. Compound Name Chemical Structure 10 Leachianone A 11 Luteolin-7-O-neohesperidoside 12 Luteolin-5-O-neohesperidoside 13 Kaempferol-7-O-α-L-arabinofuranoside 14 8-Prenylapigenin 15 Apigenine 16 Kaempferol-3-O-α-L-rhamnopyranoside OH HO O 17 Astragalin O OH OH O O OH OH OH 18 3-O-β-D-Glucopyranoside-7-O-α-L-arabinofuranoside OH HO O HO O O 19 5,7-Dihydroxychromone-7-O-β-D-glucopyranoside OH OH O Int. J. Mol. Sci.2016, 17, 2116; doi:10.3390/ijms17122116 S3 of S77 Table S1. Cont. No. Compound Name Chemical Structure 20 5,7-Dihydroxychromone-7-O-neohesperidoside Kaempferol 21 3-O-β-D-glucopyranoside-7-O-β-D-glucopyranoside 22 Xanthohumol OH HO O 23 Epicatechin OH OH OH 24 (E)-4-O-β-D-Glucopyranosyl caffeic acid 25 β-D-Glucopyranosyl sinapoic acid 26 4-O-β-D-Glucopyranosyl ferulic acid 27 Trans-caffeic acid 28 4-O-β-D-Glucopyranosyl coumaric acid 29 Dihydrocaffeic acid methyl ester 30 Dihydrocaffeic acid 31 3,4-Dihydroxyl benzoic acid 32 4-O-D-Glucosyl vanillic acid Int. -

A Meta-Analysis of the Effects of High-LET Ionizing Radiations in Human Gene Expression
Supplementary Materials A Meta-Analysis of the Effects of High-LET Ionizing Radiations in Human Gene Expression Table S1. Statistically significant DEGs (Adj. p-value < 0.01) derived from meta-analysis for samples irradiated with high doses of HZE particles, collected 6-24 h post-IR not common with any other meta- analysis group. This meta-analysis group consists of 3 DEG lists obtained from DGEA, using a total of 11 control and 11 irradiated samples [Data Series: E-MTAB-5761 and E-MTAB-5754]. Ensembl ID Gene Symbol Gene Description Up-Regulated Genes ↑ (2425) ENSG00000000938 FGR FGR proto-oncogene, Src family tyrosine kinase ENSG00000001036 FUCA2 alpha-L-fucosidase 2 ENSG00000001084 GCLC glutamate-cysteine ligase catalytic subunit ENSG00000001631 KRIT1 KRIT1 ankyrin repeat containing ENSG00000002079 MYH16 myosin heavy chain 16 pseudogene ENSG00000002587 HS3ST1 heparan sulfate-glucosamine 3-sulfotransferase 1 ENSG00000003056 M6PR mannose-6-phosphate receptor, cation dependent ENSG00000004059 ARF5 ADP ribosylation factor 5 ENSG00000004777 ARHGAP33 Rho GTPase activating protein 33 ENSG00000004799 PDK4 pyruvate dehydrogenase kinase 4 ENSG00000004848 ARX aristaless related homeobox ENSG00000005022 SLC25A5 solute carrier family 25 member 5 ENSG00000005108 THSD7A thrombospondin type 1 domain containing 7A ENSG00000005194 CIAPIN1 cytokine induced apoptosis inhibitor 1 ENSG00000005381 MPO myeloperoxidase ENSG00000005486 RHBDD2 rhomboid domain containing 2 ENSG00000005884 ITGA3 integrin subunit alpha 3 ENSG00000006016 CRLF1 cytokine receptor like -
The 1981 Significant Genes Associated with AD
Table S1: The 1981 significant genes associated with AD Uniprot Symbol Gene_name P05067 APP amyloid beta precursor protein P02649 APOE apolipoprotein E P49768 PSEN1 presenilin 1 Q92673 SORL1 sortilin related receptor 1 P10415 BCL2 BCL2, apoptosis regulator P23560 BDNF brain derived neurotrophic factor P12821 ACE angiotensin I converting enzyme P49841 GSK3B glycogen synthase kinase 3 beta P01584 IL1B interleukin 1 beta P06213 INSR insulin receptor P41159 LEP leptin P00749 PLAU plasminogen activator, urokinase P01344 IGF2 insulin like growth factor 2 P01303 NPY neuropeptide Y P08069 IGF1R insulin like growth factor 1 receptor P01308 INS insulin Q07812 BAX BCL2 associated X, apoptosis regulator P10909 CLU clusterin P49810 PSEN2 presenilin 2 Q13492 PICALM phosphatidylinositol binding clathrin assembly protein Q8IZY2 ABCA7 ATP binding cassette subfamily A member 7 Q9Y5K6 CD2AP CD2 associated protein Q9NZC2 TREM2 triggering receptor expressed on myeloid cells 2 P17927 CR1 complement C3b/C4b receptor 1 (Knops blood group) P62760 VSNL1 visinin like 1 Q9BZA7 PCDH11X protocadherin 11 X-linked Q96JQ5 MS4A4A membrane spanning 4-domains A4A P01023 A2M alpha-2-macroglobulin P22303 ACHE acetylcholinesterase (Cartwright blood group) O00499 BIN1 bridging integrator 1 P06276 BCHE butyrylcholinesterase P0DP23; CALM1 calmodulin 1 P0DP24; P0DP25 P42574 CASP3 caspase 3 P20138 CD33 CD33 molecule P36544; CHRNA7 cholinergic receptor nicotinic alpha 7 subunit Q494W8 P01034 CST3 cystatin C P10635 CYP2D6 cytochrome P450 family 2 subfamily D member 6 Q15392 DHCR24 -
1 Ligand Concentration (Μμ)
) ( 1 (51) International Patent Classification: (71) Applicant: OBSIDIAN THERAPEUTICS, INC. C12N 9/88 (2006.01) A61K 39/00 (2006.01) [US/US]; 1030 Massachusetts Avenue, Suite 400, Cam- C07K 14/705 (2006.01) bridge, MA 02138 (US). (21) International Application Number: (72) Inventors: SURI, Vipin; 3 Coolidge Road, Belmont, MA PCT/US2020/021596 02478 (US). RICHARDSON, Celeste; 199 Wolcott Road, Brookline, MA 02467 (US). DOLINSKI, Brian; c/o Ob¬ (22) International Filing Date: sidian Therapeutics, Inc., 1030 Massachusetts Avenue, 06 March 2020 (06.03.2020) Suite 400, Cambridge, MA 02138 (US). KULKARNI, Ab- (25) Filing Language: English hishek; 360 Tappan Street #3, Brookline, MA 02445 (US). INNISS, Mara Christine; 11 Highland Ave., Unit 2, Bev¬ (26) Publication Language: English erly, MA 01915 (US). SUN, Dexue; c/o Obsidian Thera¬ (30) Priority Data: peutics, Inc., 1030 Massachusetts Avenue, Suite 400, Cam¬ 62/815,399 08 March 2019 (08.03.2019) US bridge, MA 02138 (US). WEISMAN, Elizabeth Jane; c/ 62/815,402 08 March 2019 (08.03.2019) US o Obsidian Therapeutics, Inc., 1030 Massachusetts Avenue, 62/826,487 29 March 2019 (29.03.2019) US Suite 400, Cambridge, MA 02138 (US). OLINGER, Grace 62/826,443 29 March 2019 (29.03.2019) US Y.; 22 Water Street, #43 1, Cambridge, MA 02141 (US). 62/835,552 18 April 2019 (18.04.2019) US HELLER, Scott Francis; 559 Plain St., Stoughton, MA 62/835,548 18 April 2019 (18.04.2019) US 02072 (US). GORI, Jennifer Leah; 20 Spalding Street, 62/860,388 12 June 2019 (12.06.2019) US Unit 1, Jamaica Plain, MA 02130 (US). -

Choroid Plexus Genes for CSF Production and Brain Homeostasis Are Altered in Alzheimer’S Disease Shawn Kant1, Edward G
Kant et al. Fluids Barriers CNS (2018) 15:34 https://doi.org/10.1186/s12987-018-0120-7 Fluids and Barriers of the CNS RESEARCH Open Access Choroid plexus genes for CSF production and brain homeostasis are altered in Alzheimer’s disease Shawn Kant1, Edward G. Stopa1, Conrad E. Johanson2, Andrew Baird3 and Gerald D. Silverberg4* Abstract Background: The roles of the choroid plexus (CP) and cerebrospinal fuid (CSF) production have drawn increasing attention in Alzheimer’s disease (AD) research. Specifcally, studies document markedly decreased CSF production and turnover in moderate-to-severe AD. Moreover, reduced CP function and CSF turnover lead to impaired clearance of toxic metabolites, likely promote neuroinfammation, and may facilitate neuronal death during AD progression. We analyzed CP gene expression in AD compared with control subjects, specifcally considering those genes involved with CSF production and CP structural integrity. Methods: The Brown-Merck Gene Expression Omnibus (GEO) database (CP transcripts) was mined to examine changes in gene expression in AD compared to controls with a focus on assorted genes thought to play a role in CSF production. Specifcally, genes coding for ion transporters in CP epithelium (CPE) and associated enzymes like Na–K- ATPase and carbonic anhydrase, aquaporins, mitochondrial transporters/enzymes, blood–cerebrospinal fuid barrier (BCSFB) stability proteins, and pro-infammatory mediators were selected for investigation. Data were analyzed using t test p-value and fold-change analysis conducted by the GEO2R feature of the GEO database. Results: Signifcant expression changes for several genes were observed in AD CP. These included disruptions to ion transporters (e.g., the solute carrier gene SLC4A5, p 0.004) and associated enzyme expressions (e.g., carbonic anhy- drase CA4, p 0.0001), along with decreased expression= of genes involved in BCSFB integrity (e.g., claudin CLDN5, p 0.039) and= mitochondrial ATP synthesis (e.g., adenosine triphosphate ATP5L, p 0.0004). -
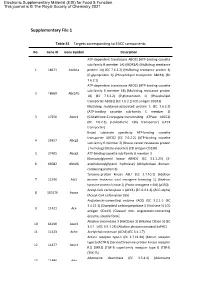
Supplementary File 1
Electronic Supplementary Material (ESI) for Food & Function. This journal is © The Royal Society of Chemistry 2021 Supplementary File 1 Table S1 Targets corresponding to EACC components No. Gene ID Gene Symbol Description ATP-dependent translocase ABCB1 (ATP-binding cassette sub-family B member 1A) (MDR1A) (Multidrug resistance 1 18671 Abcb1a protein 1A) (EC 7.6.2.2) (Multidrug resistance protein 3) (P-glycoprotein 3) (Phospholipid transporter ABCB1) (EC 7.6.2.1) ATP-dependent translocase ABCB1 (ATP-binding cassette sub-family B member 1B) (Multidrug resistance protein 2 18669 Abcb1b 1B) (EC 7.6.2.2) (P-glycoprotein 1) (Phospholipid transporter ABCB1) (EC 7.6.2.1) (CD antigen CD243) Multidrug resistance-associated protein 1 (EC 7.6.2.2) (ATP-binding cassette sub-family C member 1) 3 17250 Abcc1 (Glutathione-S-conjugate-translocating ATPase ABCC1) (EC 7.6.2.3) (Leukotriene C(4) transporter) (LTC4 transporter) Broad substrate specificity ATP-binding cassette transporter ABCG2 (EC 7.6.2.2) (ATP-binding cassette 4 26357 Abcg2 sub-family G member 2) (Breast cancer resistance protein 1 homolog) (Urate exporter) (CD antigen CD338) 5 27405 Abcg3 ATP-binding cassette sub-family G member 3 Monoacylglycerol lipase ABHD6 (EC 3.1.1.23) (2- 6 66082 Abhd6 arachidonoylglycerol hydrolase) (Abhydrolase domain- containing protein 6) Tyrosine-protein kinase ABL1 (EC 2.7.10.2) (Abelson 7 11350 Abl1 murine leukemia viral oncogene homolog 1) (Abelson tyrosine-protein kinase 1) (Proto-oncogene c-Abl) (p150) Acetyl-CoA carboxylase 1 (ACC1) (EC 6.4.1.2) (ACC-alpha)