ASPP2 Deficiency Causes Features of 1Q41q42 Microdeletion Syndrome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Universidade Estadual De Campinas Instituto De Biologia
UNIVERSIDADE ESTADUAL DE CAMPINAS INSTITUTO DE BIOLOGIA VERÔNICA APARECIDA MONTEIRO SAIA CEREDA O PROTEOMA DO CORPO CALOSO DA ESQUIZOFRENIA THE PROTEOME OF THE CORPUS CALLOSUM IN SCHIZOPHRENIA CAMPINAS 2016 1 VERÔNICA APARECIDA MONTEIRO SAIA CEREDA O PROTEOMA DO CORPO CALOSO DA ESQUIZOFRENIA THE PROTEOME OF THE CORPUS CALLOSUM IN SCHIZOPHRENIA Dissertação apresentada ao Instituto de Biologia da Universidade Estadual de Campinas como parte dos requisitos exigidos para a obtenção do Título de Mestra em Biologia Funcional e Molecular na área de concentração de Bioquímica. Dissertation presented to the Institute of Biology of the University of Campinas in partial fulfillment of the requirements for the degree of Master in Functional and Molecular Biology, in the area of Biochemistry. ESTE ARQUIVO DIGITAL CORRESPONDE À VERSÃO FINAL DA DISSERTAÇÃO DEFENDIDA PELA ALUNA VERÔNICA APARECIDA MONTEIRO SAIA CEREDA E ORIENTADA PELO DANIEL MARTINS-DE-SOUZA. Orientador: Daniel Martins-de-Souza CAMPINAS 2016 2 Agência(s) de fomento e nº(s) de processo(s): CNPq, 151787/2F2014-0 Ficha catalográfica Universidade Estadual de Campinas Biblioteca do Instituto de Biologia Mara Janaina de Oliveira - CRB 8/6972 Saia-Cereda, Verônica Aparecida Monteiro, 1988- Sa21p O proteoma do corpo caloso da esquizofrenia / Verônica Aparecida Monteiro Saia Cereda. – Campinas, SP : [s.n.], 2016. Orientador: Daniel Martins de Souza. Dissertação (mestrado) – Universidade Estadual de Campinas, Instituto de Biologia. 1. Esquizofrenia. 2. Espectrometria de massas. 3. Corpo caloso. -
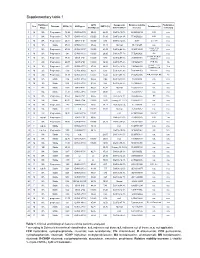
Supplementary Table 2 Supplementary Table 1
Supplementary table 1 Rai/ Binet IGHV Cytogenetic Relative viability Fludarabine- Sex Outcome CD38 (%) IGHV gene ZAP70 (%) Treatment (s) Stage identity (%) abnormalities* increase refractory 1 M 0/A Progressive 14,90 IGHV3-64*05 99,65 28,20 Del17p 18.0% 62,58322819 FCR n.a. 2 F 0/A Progressive 78,77 IGHV3-48*03 100,00 51,90 Del17p 24.8% 77,88052021 FCR n.a. 3 M 0/A Progressive 29,81 IGHV4-b*01 100,00 9,10 Del17p 12.0% 36,48 Len, Chl n.a. 4 M 1/A Stable 97,04 IGHV3-21*01 97,22 18,11 Normal 85,4191657 n.a. n.a. Chl+O, PCR, 5 F 0/A Progressive 87,00 IGHV4-39*07 100,00 43,20 Del13q 68.3% 35,23314039 n.a. HDMP+R 6 M 0/A Progressive 1,81 IGHV3-43*01 100,00 20,90 Del13q 77.7% 57,52490626 Chl n.a. Chl, FR, R-CHOP, 7 M 0/A Progressive 97,80 IGHV1-3*01 100,00 9,80 Del17p 88.5% 48,57389901 n.a. HDMP+R 8 F 2/B Progressive 69,07 IGHV5-a*03 100,00 16,50 Del17p 77.2% 107,9656878 FCR, BA No R-CHOP, FCR, 9 M 1/A Progressive 2,13 IGHV3-23*01 97,22 29,80 Del11q 16.3% 134,5866919 Yes Flavopiridol, BA 10 M 2/A Progressive 0,36 IGHV3-30*02 92,01 0,38 Del13q 81.9% 78,91844953 Unknown n.a. 11 M 2/B Progressive 15,17 IGHV3-20*01 100,00 13,20 Del11q 95.3% 75,52880995 FCR, R-CHOP, BR No 12 M 0/A Stable 0,14 IGHV3-30*02 90,62 7,40 Del13q 13.0% 13,0939004 n.a. -

CRL4-DCAF12 Ubiquitin Ligase Controls MOV10 RNA Helicase During Spermatogenesis and T Cell Activation
International Journal of Molecular Sciences Article CRL4-DCAF12 Ubiquitin Ligase Controls MOV10 RNA Helicase during Spermatogenesis and T Cell Activation Tomas Lidak 1,2, Nikol Baloghova 1, Vladimir Korinek 1,3 , Radislav Sedlacek 4, Jana Balounova 4 , Petr Kasparek 4 and Lukas Cermak 1,* 1 Laboratory of Cancer Biology, Institute of Molecular Genetics of the Czech Academy of Sciences, 252 42 Vestec, Czech Republic; [email protected] (T.L.); [email protected] (N.B.); [email protected] (V.K.) 2 Faculty of Science, Charles University, 128 00 Prague, Czech Republic 3 Laboratory of Cell and Developmental Biology, Institute of Molecular Genetics of the Czech Academy of Sciences, 252 42 Vestec, Czech Republic 4 Czech Centre for Phenogenomics, Institute of Molecular Genetics of the Czech Academy of Sciences, 252 50 Vestec, Czech Republic; [email protected] (R.S.); [email protected] (J.B.); [email protected] (P.K.) * Correspondence: [email protected] Abstract: Multisubunit cullin-RING ubiquitin ligase 4 (CRL4)-DCAF12 recognizes the C-terminal degron containing acidic amino acid residues. However, its physiological roles and substrates are largely unknown. Purification of CRL4-DCAF12 complexes revealed a wide range of potential substrates, including MOV10, an “ancient” RNA-induced silencing complex (RISC) complex RNA helicase. We show that DCAF12 controls the MOV10 protein level via its C-terminal motif in a Citation: Lidak, T.; Baloghova, N.; proteasome- and CRL-dependent manner. Next, we generated Dcaf12 knockout mice and demon- Korinek, V.; Sedlacek, R.; Balounova, strated that the DCAF12-mediated degradation of MOV10 is conserved in mice and humans. -

The UVB-Induced Gene Expression Profile of Human Epidermis in Vivo Is Different from That of Cultured Keratinocytes
Oncogene (2006) 25, 2601–2614 & 2006 Nature Publishing Group All rights reserved 0950-9232/06 $30.00 www.nature.com/onc ORIGINAL ARTICLE The UVB-induced gene expression profile of human epidermis in vivo is different from that of cultured keratinocytes CD Enk1, J Jacob-Hirsch2, H Gal3, I Verbovetski4, N Amariglio2, D Mevorach4, A Ingber1, D Givol3, G Rechavi2 and M Hochberg1 1Department of Dermatology, The Hadassah-Hebrew University Medical Center, Jerusalem, Israel; 2Department of Pediatric Hemato-Oncology and Functional Genomics, Safra Children’s Hospital, Sheba Medical Center and Sackler School of Medicine, Tel-Aviv University,Tel Aviv, Israel; 3Department of Molecular Cell Biology, Weizmann Institute of Science, Rehovot, Israel and 4The Laboratory for Cellular and Molecular Immunology, Department of Medicine, The Hadassah-Hebrew University Medical Center, Jerusalem, Israel In order to obtain a comprehensive picture of the radiation. UVB, with a wavelength range between 290 molecular events regulating cutaneous photodamage of and 320 nm, represents one of the most important intact human epidermis, suction blister roofs obtained environmental hazards affectinghuman skin (Hahn after a single dose of in vivo ultraviolet (UV)B exposure and Weinberg, 2002). To protect itself against the were used for microarray profiling. We found a changed DNA-damaging effects of sunlight, the skin disposes expression of 619 genes. Half of the UVB-regulated genes over highly complicated cellular programs, including had returned to pre-exposure baseline levels at 72 h, cell-cycle arrest, DNA repair and apoptosis (Brash et al., underscoring the transient character of the molecular 1996). Failure in selected elements of these defensive cutaneous UVB response. -

Investigation of the Underlying Hub Genes and Molexular Pathogensis in Gastric Cancer by Integrated Bioinformatic Analyses
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.20.423656; this version posted December 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Investigation of the underlying hub genes and molexular pathogensis in gastric cancer by integrated bioinformatic analyses Basavaraj Vastrad1, Chanabasayya Vastrad*2 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2020.12.20.423656; this version posted December 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract The high mortality rate of gastric cancer (GC) is in part due to the absence of initial disclosure of its biomarkers. The recognition of important genes associated in GC is therefore recommended to advance clinical prognosis, diagnosis and and treatment outcomes. The current investigation used the microarray dataset GSE113255 RNA seq data from the Gene Expression Omnibus database to diagnose differentially expressed genes (DEGs). Pathway and gene ontology enrichment analyses were performed, and a proteinprotein interaction network, modules, target genes - miRNA regulatory network and target genes - TF regulatory network were constructed and analyzed. Finally, validation of hub genes was performed. The 1008 DEGs identified consisted of 505 up regulated genes and 503 down regulated genes. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Study on Apoptosis in Human Deciduous Tooth Pulp Cells Original
Yuko Okai et al.: Apoptosis in Human Deciduous Tooth Pulp Cells Journal of Hard Tissue Biology 21[4] (2012) p413-420 © 2012 The Hard Tissue Biology Network Association Printed in Japan, All rights reserved. CODEN-JHTBFF, ISSN 1341-7649 Original Study on Apoptosis in Human Deciduous Tooth Pulp Cells Yuko Okai1), Kyoko Harada1), Kiyoshi Ohura2) and Kenji Arita1) 1)Department of Pediatric Dentistry, Osaka Dental University,Osaka, Japan 2)Department of Pharmacology, Osaka Dental University, Osaka, Japan (Accepted for publication, July 5, 2012) Abstract: Dental pulp plays an important role in tooth vitality. The pulp of deciduous tooth thus shows vigorous metabolism and repeated, vigorous cell regeneration and proliferation. Furthermore, natural, programmed cell death (apoptosis) frequently occurs as part of this cell regeneration and proliferation process. Apoptosis is defined as cell death governed by intracellular signals (active death of the cell triggered by changes in physiological or external conditions), and the birth of new cells occurs simultaneously with cell death. Apoptosis thus functions as a driving force for morphological and other changes in organisms, and is essential for phenomena such as ontogenetic processes or aging. However, In order to clarify the mechanism of apoptosis in the deciduous tooth pulp cells, we used SuperArray analysis and immunostaining to examine both deciduous and permanent tooth pulp cells. Of the 84 major genes involved in apoptosis, seven showed greater expression in the deciduous tooth pulp cells than in the permanent tooth pulp cells. Gene expression of TP53BP2, CRADD, BAK1, BIRC3, CASP8, CASP2 and TP73 in the deciduous tooth pulp were 16.1, 9.0, 8.5, 8.1, 5.9, 5.8 and 3.2 times greater than those in the permanent tooth pulp cells, respectively. -

Inhibition of Mitochondrial Complex II in Neuronal Cells Triggers Unique
www.nature.com/scientificreports OPEN Inhibition of mitochondrial complex II in neuronal cells triggers unique pathways culminating in autophagy with implications for neurodegeneration Sathyanarayanan Ranganayaki1, Neema Jamshidi2, Mohamad Aiyaz3, Santhosh‑Kumar Rashmi4, Narayanappa Gayathri4, Pulleri Kandi Harsha5, Balasundaram Padmanabhan6 & Muchukunte Mukunda Srinivas Bharath7* Mitochondrial dysfunction and neurodegeneration underlie movement disorders such as Parkinson’s disease, Huntington’s disease and Manganism among others. As a corollary, inhibition of mitochondrial complex I (CI) and complex II (CII) by toxins 1‑methyl‑4‑phenylpyridinium (MPP+) and 3‑nitropropionic acid (3‑NPA) respectively, induced degenerative changes noted in such neurodegenerative diseases. We aimed to unravel the down‑stream pathways associated with CII inhibition and compared with CI inhibition and the Manganese (Mn) neurotoxicity. Genome‑wide transcriptomics of N27 neuronal cells exposed to 3‑NPA, compared with MPP+ and Mn revealed varied transcriptomic profle. Along with mitochondrial and synaptic pathways, Autophagy was the predominant pathway diferentially regulated in the 3‑NPA model with implications for neuronal survival. This pathway was unique to 3‑NPA, as substantiated by in silico modelling of the three toxins. Morphological and biochemical validation of autophagy markers in the cell model of 3‑NPA revealed incomplete autophagy mediated by mechanistic Target of Rapamycin Complex 2 (mTORC2) pathway. Interestingly, Brain Derived Neurotrophic Factor -

Hsa-Mir-21-5P
Supporting Information An exosomal urinary miRNA signature for early diagnosis of renal fibrosis in Lupus Nephritis 1 2 2 1 1 Cristina Solé , Teresa Moliné , Marta Vidal , Josep Ordi-Ros , Josefina Cortés-Hernández 1Hospital Universitari Vall d’Hebron, Vall d’Hebron Research Institute (VHIR), Lupus Unit, Barcelona, Spain; [email protected] (C.S); [email protected] (J.O-R.); [email protected] (J.C-H.) 2 Hospital Universitari Vall d’Hebron, Department of Renal Pathology, Barcelona, Spain; [email protected] (T.M); [email protected] (M.V). INDEX 1. SI Materials and Methods......................................................................................3 2. SI References…………………………………………………………………………….7 2. SI Figures................................................................................................................8 4. SI Tables................................................................................................................21 2 SI Materials and Methods Patients and samples Patients with biopsy-proven active LN were recruited from the Lupus Unit at Vall d’Hebron Hospital (N=45). All patients fulfilled at least 4 of the American College of rheumatology (ACR) revised classification criteria for SLE [1]. Healthy donors were used as controls (N=20). Urine samples were collected from each patient 1 day before renal biopsy and processed immediately to be stored at -80ºC. Patients with urinary tract infection, diabetes mellitus, pregnancy, malignancy and non-lupus-related renal failure were excluded. In addition, key laboratory measurements were obtained including complement levels (C3 and C4), anti-double-stranded DNA (anti-dsDNA), 24-h proteinuria, blood urea nitrogen (BUN), serum creatinine and the estimated glomerular filtration ratio (eGFR) using the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) formula [2]. SLE disease activity was assessed by the SLE Disease Activity Index 2000 update (SLEDAI-2Ks; range 0–105) [3]. -

Human Lectins, Their Carbohydrate Affinities and Where to Find Them
biomolecules Review Human Lectins, Their Carbohydrate Affinities and Where to Review HumanFind Them Lectins, Their Carbohydrate Affinities and Where to FindCláudia ThemD. Raposo 1,*, André B. Canelas 2 and M. Teresa Barros 1 1, 2 1 Cláudia D. Raposo * , Andr1 é LAQVB. Canelas‐Requimte,and Department M. Teresa of Chemistry, Barros NOVA School of Science and Technology, Universidade NOVA de Lisboa, 2829‐516 Caparica, Portugal; [email protected] 12 GlanbiaLAQV-Requimte,‐AgriChemWhey, Department Lisheen of Chemistry, Mine, Killoran, NOVA Moyne, School E41 of ScienceR622 Co. and Tipperary, Technology, Ireland; canelas‐ [email protected] NOVA de Lisboa, 2829-516 Caparica, Portugal; [email protected] 2* Correspondence:Glanbia-AgriChemWhey, [email protected]; Lisheen Mine, Tel.: Killoran, +351‐212948550 Moyne, E41 R622 Tipperary, Ireland; [email protected] * Correspondence: [email protected]; Tel.: +351-212948550 Abstract: Lectins are a class of proteins responsible for several biological roles such as cell‐cell in‐ Abstract:teractions,Lectins signaling are pathways, a class of and proteins several responsible innate immune for several responses biological against roles pathogens. such as Since cell-cell lec‐ interactions,tins are able signalingto bind to pathways, carbohydrates, and several they can innate be a immuneviable target responses for targeted against drug pathogens. delivery Since sys‐ lectinstems. In are fact, able several to bind lectins to carbohydrates, were approved they by canFood be and a viable Drug targetAdministration for targeted for drugthat purpose. delivery systems.Information In fact, about several specific lectins carbohydrate were approved recognition by Food by andlectin Drug receptors Administration was gathered for that herein, purpose. plus Informationthe specific organs about specific where those carbohydrate lectins can recognition be found by within lectin the receptors human was body. -

Modes of Cell Death Induced by Photodynamic Therapy Using Zinc Phthalocyanine in Lung Cancer Cells Grown As a Monolayer and Three-Dimensional Multicellular Spheroids
molecules Article Modes of Cell Death Induced by Photodynamic Therapy Using Zinc Phthalocyanine in Lung Cancer Cells Grown as a Monolayer and Three-Dimensional Multicellular Spheroids Sello L. Manoto, Nicolette Houreld, Natasha Hodgkinson and Heidi Abrahamse * Laser Research Centre, Faculty of Health Sciences, University of Johannesburg, P.O. Box 17011, Doornfontein 2028, South Africa; [email protected] (S.L.M.); [email protected] (N.H.); [email protected] (N.H.) * Correspondence: [email protected]; Tel.: +27-11-559-6406; Fax: +27-11-559-6884 Academic Editors: Augusto C. Tomé, João Paulo C. Tomé and Michael Hanack Received: 8 March 2017; Accepted: 9 May 2017; Published: 16 May 2017 Abstract: Photodynamic therapy (PDT) involves interaction of a photosensitizer, light, and molecular oxygen which produces singlet oxygen and subsequent tumour eradication. The development of second generation photosensitizers, such as phthalocyanines, has improved this technology. Customary monolayer cell culture techniques are, unfortunately, too simple to replicate treatment effects in vivo. Multicellular tumour spheroids may provide a better alternative since they mimic aspects of the human tumour environment. This study aimed to profile 84 genes involved in apoptosis following treatment with PDT on lung cancer cells (A549) grown in a monolayer versus three-dimensional multicellular tumour spheroids (250 and 500 µm). Gene expression profiling 2 was performed 24 h post irradiation (680 nm; 5 J/cm ) with zinc sulfophthalocyanine (ZnPcSmix) to determine the genes involved in apoptotic cell death. In the monolayer cells, eight pro-apoptotic genes were upregulated, and two were downregulated. In the multicellular tumour spheroids (250 µm) there was upregulation of only 1 gene while there was downregulation of 56 genes. -

A Four-Gene Decision Tree Signature
Published OnlineFirst October 10, 2018; DOI: 10.1158/1535-7163.MCT-18-0243 Companion Diagnostic, Pharmacogenomic, and Cancer Biomarkers Molecular Cancer Therapeutics A Four-gene Decision Tree Signature Classification of Triple-negative Breast Cancer: Implications for Targeted Therapeutics Jelmar Quist1,2,3, Hasan Mirza1,2,3, Maggie C.U. Cheang4,5, Melinda L. Telli6, Joyce A. O'Shaughnessy7, Christopher J. Lord5,8, Andrew N.J. Tutt2,3,5, and Anita Grigoriadis1,2,3 Abstract The molecular complexity of triple-negative breast cancers in early diagnosed TNBCs. In TNBC patients with metastatic (TNBCs) provides a challenge for patient management. We set disease, the MC6 proportion was reduced to 25%, and was out to characterize this heterogeneous disease by combining independently associated with a higher response rate to transcriptomics and genomics data, with the aim of revealing platinum-based chemotherapy. In TNBC cell line data, convergent pathway dependencies with the potential for treat- platinum sensitivity was recapitulated, and a sensitivity ment intervention. A Bayesian algorithm was used to integrate to the inhibition of the phosphatase PPM1D was revealed. molecular profiles in two TNBC cohorts, followed by valida- Molecularly, MC6-TNBCs displayed high levels of telomeric þ þ tion using five independent cohorts (n ¼ 1,168), including allelic imbalances, enrichment of CD4 and CD8 immune three clinical trials. A four-gene decision tree signature was signatures, and reduced expression of genes negatively reg- identified, which robustly classified TNBCs into six subtypes. ulating the MAPK signaling pathway. These observations All four genes in the signature (EXO1, TP53BP2, FOXM1, and suggest that our integrative classification approach may RSU1) are associated with either genomic instability, malig- identify TNBC patients with discernible and theoretically nant growth, or treatment response.