A 15Q25.2 Microdeletion Phenotype For
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Trans Esnps Infers Regulatory Network Architecture
Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2014 © 2014 Anat Kreimer All rights reserved ABSTRACT Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer eSNPs are genetic variants associated with transcript expression levels. The characteristics of such variants highlight their importance and present a unique opportunity for studying gene regulation. eSNPs affect most genes and their cell type specificity can shed light on different processes that are activated in each cell. They can identify functional variants by connecting SNPs that are implicated in disease to a molecular mechanism. Examining eSNPs that are associated with distal genes can provide insights regarding the inference of regulatory networks but also presents challenges due to the high statistical burden of multiple testing. Such association studies allow: simultaneous investigation of many gene expression phenotypes without assuming any prior knowledge and identification of unknown regulators of gene expression while uncovering directionality. This thesis will focus on such distal eSNPs to map regulatory interactions between different loci and expose the architecture of the regulatory network defined by such interactions. We develop novel computational approaches and apply them to genetics-genomics data in human. We go beyond pairwise interactions to define network motifs, including regulatory modules and bi-fan structures, showing them to be prevalent in real data and exposing distinct attributes of such arrangements. We project eSNP associations onto a protein-protein interaction network to expose topological properties of eSNPs and their targets and highlight different modes of distal regulation. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Circular RNA Hsa Circ 0005114‑Mir‑142‑3P/Mir‑590‑5P‑ Adenomatous
ONCOLOGY LETTERS 21: 58, 2021 Circular RNA hsa_circ_0005114‑miR‑142‑3p/miR‑590‑5p‑ adenomatous polyposis coli protein axis as a potential target for treatment of glioma BO WEI1*, LE WANG2* and JINGWEI ZHAO1 1Department of Neurosurgery, China‑Japan Union Hospital of Jilin University, Changchun, Jilin 130033; 2Department of Ophthalmology, The First Hospital of Jilin University, Jilin University, Changchun, Jilin 130021, P.R. China Received September 12, 2019; Accepted October 22, 2020 DOI: 10.3892/ol.2020.12320 Abstract. Glioma is the most common type of brain tumor APC expression with a good overall survival rate. UALCAN and is associated with a high mortality rate. Despite recent analysis using TCGA data of glioblastoma multiforme and the advances in treatment options, the overall prognosis in patients GSE25632 and GSE103229 microarray datasets showed that with glioma remains poor. Studies have suggested that circular hsa‑miR‑142‑3p/hsa‑miR‑590‑5p was upregulated and APC (circ)RNAs serve important roles in the development and was downregulated. Thus, hsa‑miR‑142‑3p/hsa‑miR‑590‑5p‑ progression of glioma and may have potential as therapeutic APC‑related circ/ceRNA axes may be important in glioma, targets. However, the expression profiles of circRNAs and their and hsa_circ_0005114 interacted with both of these miRNAs. functions in glioma have rarely been studied. The present study Functional analysis showed that hsa_circ_0005114 was aimed to screen differentially expressed circRNAs (DECs) involved in insulin secretion, while APC was associated with between glioma and normal brain tissues using sequencing the Wnt signaling pathway. In conclusion, hsa_circ_0005114‑ data collected from the Gene Expression Omnibus database miR‑142‑3p/miR‑590‑5p‑APC ceRNA axes may be potential (GSE86202 and GSE92322 datasets) and explain their mecha‑ targets for the treatment of glioma. -
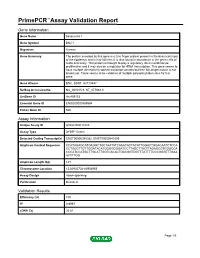
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name basonuclin 1 Gene Symbol BNC1 Organism Human Gene Summary The protein encoded by this gene is a zinc finger protein present in the basal cell layer of the epidermis and in hair follicles. It is also found in abundance in the germ cells of testis and ovary. This protein is thought to play a regulatory role in keratinocyte proliferation and it may also be a regulator for rRNA transcription. This gene seems to have multiple alternatively spliced transcript variants but their full-length nature is not known yet. There seems to be evidence of multiple polyadenylation sites for this gene. Gene Aliases BNC, BSN1, HsT19447 RefSeq Accession No. NC_000015.9, NT_077661.3 UniGene ID Hs.459153 Ensembl Gene ID ENSG00000169594 Entrez Gene ID 646 Assay Information Unique Assay ID qHsaCID0017223 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000345382, ENST00000541809 Amplicon Context Sequence CCATAGAGCATGAGGCTGCTAATATCAAACACTACATTGGACTGGACAATCTCCA CCTGGCTTGTTGGATACATGGGGGGGATCCTTAGCTTACTTAGAGCGTGGGCCA CCCATCCATGCTTGCATTGGTCACACTGACGGTGGTTTATTTTCCCGGGTTTGAA ACTTTGG Amplicon Length (bp) 141 Chromosome Location 15:83935724-83936955 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 100 R2 0.9997 cDNA Cq 26.81 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 84.5 gDNA Cq 39.42 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report BNC1, Human Amplification -

Mitotic Chromosome Binding Predicts Transcription Factor Properties in Interphase
bioRxiv preprint doi: https://doi.org/10.1101/404723; this version posted August 31, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Mitotic chromosome binding predicts transcription factor properties in interphase Authors: Mahé Raccaud1, Andrea B. Alber1,2, Elias T. Friman1,2, Harsha Agarwal3, Cédric Deluz1, Timo Kuhn3, J. Christof M. Gebhardt3 and David M. Suter1,4 Affiliations: 1Institute of Bioengineering, School of Life Sciences, Ecole Polytechnique Fédérale de Lausanne (EPFL), Lausanne, CH-1015, Switzerland 2These authors contributed equally 3Institute of Biophysics, Ulm University, Albert-Einstein-Allee 11, Ulm 89081, Germany 4Lead contact Corresponding author: David M. Suter ([email protected]) Summary Mammalian transcription factors (TFs) differ broadly in their nuclear mobility and sequence- specific/non-specific DNA binding affinity. How these properties affect the ability of TFs to occupy their specific binding sites in the genome and modify the epigenetic landscape is unclear. Here we combined live cell quantitative measurements of mitotic chromosome binding (MCB) of 502 TFs, measurements of TF mobility by fluorescence recovery after photobleaching, single molecule imaging of DNA binding in live cells, and genome-wide mapping of TF binding and chromatin accessibility. MCB scaled with interphase properties such as association with DNA-rich compartments, mobility, as well as large differences in genome-wide specific site occupancy that correlated with TF impact on chromatin accessibility. As MCB is largely mediated by electrostatic, non-specific TF-DNA interactions, our data suggests that non-specific DNA binding of TFs enhances their search for specific sites and thereby their impact on the accessible chromatin landscape. -

The DNA Methylation Landscape of Glioblastoma Disease Progression Shows Extensive Heterogeneity in Time and Space
bioRxiv preprint doi: https://doi.org/10.1101/173864; this version posted August 9, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space Johanna Klughammer1*, Barbara Kiesel2,3*, Thomas Roetzer3,4, Nikolaus Fortelny1, Amelie Kuchler1, Nathan C. Sheffield5, Paul Datlinger1, Nadine Peter3,4, Karl-Heinz Nenning6, Julia Furtner3,7, Martha Nowosielski8,9, Marco Augustin10, Mario Mischkulnig2,3, Thomas Ströbel3,4, Patrizia Moser11, Christian F. Freyschlag12, Jo- hannes Kerschbaumer12, Claudius Thomé12, Astrid E. Grams13, Günther Stockhammer8, Melitta Kitzwoegerer14, Stefan Oberndorfer15, Franz Marhold16, Serge Weis17, Johannes Trenkler18, Johanna Buchroithner19, Josef Pichler20, Johannes Haybaeck21,22, Stefanie Krassnig21, Kariem Madhy Ali23, Gord von Campe23, Franz Payer24, Camillo Sherif25, Julius Preiser26, Thomas Hauser27, Peter A. Winkler27, Waltraud Kleindienst28, Franz Würtz29, Tanisa Brandner-Kokalj29, Martin Stultschnig30, Stefan Schweiger31, Karin Dieckmann3,32, Matthias Preusser3,33, Georg Langs6, Bernhard Baumann10, Engelbert Knosp2,3, Georg Widhalm2,3, Christine Marosi3,33, Johannes A. Hainfellner3,4, Adelheid Woehrer3,4#§, Christoph Bock1,34,35# 1 CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences, Vienna, Austria. 2 Department of Neurosurgery, Medical University of Vienna, Vienna, Austria. 3 Comprehensive Cancer Center, Central Nervous System Tumor Unit, Medical University of Vienna, Austria. 4 Institute of Neurology, Medical University of Vienna, Vienna, Austria. 5 Center for Public Health Genomics, University of Virginia, Charlottesville VA, USA. 6 Department of Biomedical Imaging and Image-guided Therapy, Computational Imaging Research Lab, Medical University of Vi- enna, Vienna, Austria. -

Identification of Novel DNA-Methylated Genes
Prostate Cancer and Prostatic Disease (2013) 16, 292–300 & 2013 Macmillan Publishers Limited All rights reserved 1365-7852/13 www.nature.com/pcan ORIGINAL ARTICLE Identification of novel DNA-methylated genes that correlate with human prostate cancer and high-grade prostatic intraepithelial neoplasia JM Devaney1, S Wang2,3, S Funda1, J Long2, DJ Taghipour2, R Tbaishat3, P Furbert-Harris2,4, M Ittmann5 and B Kwabi-Addo2,3 BACKGROUND: Prostate cancer (PCa) harbors a myriad of genomic and epigenetic defects. Cytosine methylation of CpG-rich promoter DNA is an important mechanism of epigenetic gene inactivation in PCa. There is considerable amount of data to suggest that DNA methylation-based biomarkers may be useful for the early detection and diagnosis of PCa. In addition, candidate gene- based studies have shown an association between specific gene methylation and alterations and clinicopathologic indicators of poor prognosis in PCa. METHODS: To more comprehensively identify DNA methylation alterations in PCa initiation and progression, we examined the methylation status of 485 577 CpG sites from regions with a broad spectrum of CpG densities, interrogating both gene-associated and non-associated regions using the recently developed Illumina 450K methylation platform. RESULTS: In all, we selected 33 promoter-associated novel CpG sites that were differentially methylated in high-grade prostatic intraepithelial neoplasia and PCa in comparison with benign prostate tissue samples (false discovery rate-adjusted P-value o0.05; b-value X0.2; fold change 41.5). Of the 33 genes, hierarchical clustering analysis demonstrated BNC1, FZD1, RPL39L, SYN2, LMX1B, CXXC5, ZNF783 and CYB5R2 as top candidate novel genes that are frequently methylated and whose methylation was associated with inactivation of gene expression in PCa cell lines. -

Noelia Díaz Blanco
Effects of environmental factors on the gonadal transcriptome of European sea bass (Dicentrarchus labrax), juvenile growth and sex ratios Noelia Díaz Blanco Ph.D. thesis 2014 Submitted in partial fulfillment of the requirements for the Ph.D. degree from the Universitat Pompeu Fabra (UPF). This work has been carried out at the Group of Biology of Reproduction (GBR), at the Department of Renewable Marine Resources of the Institute of Marine Sciences (ICM-CSIC). Thesis supervisor: Dr. Francesc Piferrer Professor d’Investigació Institut de Ciències del Mar (ICM-CSIC) i ii A mis padres A Xavi iii iv Acknowledgements This thesis has been made possible by the support of many people who in one way or another, many times unknowingly, gave me the strength to overcome this "long and winding road". First of all, I would like to thank my supervisor, Dr. Francesc Piferrer, for his patience, guidance and wise advice throughout all this Ph.D. experience. But above all, for the trust he placed on me almost seven years ago when he offered me the opportunity to be part of his team. Thanks also for teaching me how to question always everything, for sharing with me your enthusiasm for science and for giving me the opportunity of learning from you by participating in many projects, collaborations and scientific meetings. I am also thankful to my colleagues (former and present Group of Biology of Reproduction members) for your support and encouragement throughout this journey. To the “exGBRs”, thanks for helping me with my first steps into this world. Working as an undergrad with you Dr. -

Multivariate Analysis Reveals Genetic Associations of the Resting Default
Multivariate analysis reveals genetic associations of PNAS PLUS the resting default mode network in psychotic bipolar disorder and schizophrenia Shashwath A. Medaa,1, Gualberto Ruañob,c, Andreas Windemuthb, Kasey O’Neila, Clifton Berwisea, Sabra M. Dunna, Leah E. Boccaccioa, Balaji Narayanana, Mohan Kocherlab, Emma Sprootena, Matcheri S. Keshavand, Carol A. Tammingae, John A. Sweeneye, Brett A. Clementzf, Vince D. Calhoung,h,i, and Godfrey D. Pearlsona,h,j aOlin Neuropsychiatry Research Center, Institute of Living at Hartford Hospital, Hartford, CT 06102; bGenomas Inc., Hartford, CT 06102; cGenetics Research Center, Hartford Hospital, Hartford, CT 06102; dDepartment of Psychiatry, Beth Israel Deaconess Hospital, Harvard Medical School, Boston, MA 02215; eDepartment of Psychiatry, University of Texas Southwestern Medical Center, Dallas, TX 75390; fDepartment of Psychology, University of Georgia, Athens, GA 30602; gThe Mind Research Network, Albuquerque, NM 87106; Departments of hPsychiatry and jNeurobiology, Yale University, New Haven, CT 06520; and iDepartment of Electrical and Computer Engineering, The University of New Mexico, Albuquerque, NM 87106 Edited by Robert Desimone, Massachusetts Institute of Technology, Cambridge, MA, and approved April 4, 2014 (received for review July 15, 2013) The brain’s default mode network (DMN) is highly heritable and is Although risk for psychotic illnesses is driven in small part by compromised in a variety of psychiatric disorders. However, ge- highly penetrant, often private mutations such as copy number netic control over the DMN in schizophrenia (SZ) and psychotic variants, substantial risk also is likely conferred by multiple genes bipolar disorder (PBP) is largely unknown. Study subjects (n = of small effect sizes interacting together (7). According to the 1,305) underwent a resting-state functional MRI scan and were “common disease common variant” (CDCV) model, one would analyzed by a two-stage approach. -

Structural Variant Calling by Assembly in Whole Human Genomes: Applications in Hypoplastic Left Heart Syndrome by Matthew Kendzi
STRUCTURAL VARIANT CALLING BY ASSEMBLY IN WHOLE HUMAN GENOMES: APPLICATIONS IN HYPOPLASTIC LEFT HEART SYNDROME BY MATTHEW KENDZIOR THESIS Submitted in partial FulFillment oF tHe requirements for the degree of Master of Science in BioinFormatics witH a concentration in Crop Sciences in the Graduate College of the University oF Illinois at Urbana-Champaign, 2019 Urbana, Illinois Master’s Committee: ProFessor MattHew Hudson, CHair ResearcH Assistant ProFessor Liudmila Mainzer ProFessor SaurabH SinHa ABSTRACT Variant discovery in medical researcH typically involves alignment oF sHort sequencing reads to the human reference genome. SNPs and small indels (variants less than 50 nucleotides) are the most common types oF variants detected From alignments. Structural variation can be more diFFicult to detect From short-read alignments, and thus many software applications aimed at detecting structural variants From short read alignments have been developed. However, these almost all detect the presence of variation in a sample using expected mate-pair distances From read data, making them unable to determine the precise sequence of the variant genome at the speciFied locus. Also, reads from a structural variant allele migHt not even map to the reference, and will thus be lost during variant discovery From read alignment. A variant calling by assembly approacH was used witH tHe soFtware Cortex-var for variant discovery in Hypoplastic Left Heart Syndrome (HLHS). THis method circumvents many of the limitations oF variants called From a reFerence alignment: unmapped reads will be included in a sample’s assembly, and variants up to thousands of nucleotides can be detected, with the Full sample variant allele sequence predicted. -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine