Enzyme Evolution: Exploring Substrate Scope and Catalytic Promiscuity
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

(12) United States Patent (10) Patent No.: US 8,530,724 B2 Whitelaw Et Al
US008530724B2 (12) United States Patent (10) Patent No.: US 8,530,724 B2 Whitelaw et al. (45) Date of Patent: Sep. 10, 2013 (54) ALTERING THE FATTY ACID COMPOSITION 2011/0054198 A1 3/2011 Singh et al. OF RICE 2011/O190521 A1 8/2011 Damcevski et al. 2011/02O1065 A1 8/2011 Singh et al. 2011/02233.11 A1 9, 2011 Liu et al. (75) Inventors: Ella Whitelaw, Albury (AU); Sadequr 2011/0229623 A1 9, 2011 Liu et al. Rahman, Nicholls (AU); Zhongyi Li, 2012/0041218 A1 2/2012 Singh et al. Kaleen (AU); Qing Liu, Girralang (AU); Surinder Pal Singh, Downer (AU): FOREIGN PATENT DOCUMENTS WO WO99,049050 9, 1999 Robert Charles de Feyter, Monash WO WOO3,080802 10, 2003 (AU) WO WO 2003/080802 A2 10/2003 WO WO 2004/001001 12/2003 (73) Assignee: Commonwealth Scientific and WO WO 2004/001001 A2 12/2003 Industrial Research Organisation, WO WO 2008/O25068 6, 2008 Campbell (AU) WO WO 2009/12958 10/2009 WO WO 2010/009:500 1, 2010 (*) Notice: Subject to any disclaimer, the term of this WO WO 2010/057246 5, 2010 patent is extended or adjusted under 35 OTHER PUBLICATIONS U.S.C. 154(b) by 772 days. Supplementary European Search Report issued Feb. 23, 2010 in connection with corresponding European Patent Application No. (21) Appl. No.: 12/309.276 0776.37759. Taira et al. (1989) “Fatty Acid Composition of Indica-Types and (22) PCT Filed: Jul. 13, 2007 Japonica-Types of Rice Bran and Milled Rice' Journal of the Ameri can Oil Chemists’ Society; vol. -

Rhodococcus Erythropolis Oleate Hydratase: a New Member in the Oleate Hydratase Family Tree – Biochemical and Structural Studies
Technische Universität München Fakultät für Chemie Werner Siemens-Lehrstuhl für Synthetische Biotechnologie Enzymatic functionalization of bio based fatty acids and algae based triglycerides Jan Lorenzen Vollständiger Abdruck der von der Fakultät für Chemie der Technischen Universität München zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.) genehmigten Dissertation. Vorsitzender Prof. Dr. Tom Nilges Prüfer der Dissertation 1. Prof. Dr. Thomas Brück 2. Prof. Dr. Wolfgang Eisenreich 3. Prof. Dr. Uwe Bornscheuer (schriftliche Beurteilung) Prof. Dr. Thomas Fässler (mündliche Prüfung) Die Dissertation wurde am 26.09.2019 bei der Technischen Universität München eingereicht und durch die Fakultät für Chemie am 19.11.2019 angenommen. Abstracts I Abstracts Chapter I In the first chapter, the optimization of the SCCO2 extraction of microalgae derived biomass in an industrially relevant pilot plant that adheres to ATEX standards was reported for the first time. The SCCO2 extraction procedure of the lipid containing biomass of the microalgae strains Scenedesmus obliquus and Scenedesmus obtusiusculus was optimized with batch sizes of up to 1,3 kg of dried biomass. Under optimal conditions lipid recovery up to 92% w/w of the total lipids fraction could be achieved. Moreover, we addressed the purification of crude microalgae lipid extracts obtained by the SCCO2 extraction procedure. We could identify high amounts of hydrophobic contaminants (e.g. chlorophylls or carotenoids) in the crude lipid extract, which we could effectively remove by the application of the standard mineral adsorber bentonite. The obtained, clean microalgae oil can directly be further processed to lubricants, cosmetics or nutraceutical products. This is the first time that a combined SCCO2 extraction and purification process for algae lipids has been developed that could be integrated into existing industrial processes. -

Supplementary Table 9. Functional Annotation Clustering Results for the Union (GS3) of the Top Genes from the SNP-Level and Gene-Based Analyses (See ST4)
Supplementary Table 9. Functional Annotation Clustering Results for the union (GS3) of the top genes from the SNP-level and Gene-based analyses (see ST4) Column Header Key Annotation Cluster Name of cluster, sorted by descending Enrichment score Enrichment Score EASE enrichment score for functional annotation cluster Category Pathway Database Term Pathway name/Identifier Count Number of genes in the submitted list in the specified term % Percentage of identified genes in the submitted list associated with the specified term PValue Significance level associated with the EASE enrichment score for the term Genes List of genes present in the term List Total Number of genes from the submitted list present in the category Pop Hits Number of genes involved in the specified term (category-specific) Pop Total Number of genes in the human genome background (category-specific) Fold Enrichment Ratio of the proportion of count to list total and population hits to population total Bonferroni Bonferroni adjustment of p-value Benjamini Benjamini adjustment of p-value FDR False Discovery Rate of p-value (percent form) Annotation Cluster 1 Enrichment Score: 3.8978262119731335 Category Term Count % PValue Genes List Total Pop Hits Pop Total Fold Enrichment Bonferroni Benjamini FDR GOTERM_CC_DIRECT GO:0005886~plasma membrane 383 24.33290978 5.74E-05 SLC9A9, XRCC5, HRAS, CHMP3, ATP1B2, EFNA1, OSMR, SLC9A3, EFNA3, UTRN, SYT6, ZNRF2, APP, AT1425 4121 18224 1.18857065 0.038655922 0.038655922 0.086284383 UP_KEYWORDS Membrane 626 39.77128335 1.53E-04 SLC9A9, HRAS, -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota. -

(12) Patent Application Publication (10) Pub. No.: US 2015/0037860 A1 B0tes Et Al
US 2015 0037860A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2015/0037860 A1 B0tes et al. (43) Pub. Date: Feb. 5, 2015 (54) METHODS FOR BOSYNTHESIS OF Publication Classification ISOPRENE (51) Int. C. (71) Applicant: INVISTA North America S.ár.l., CI2P 5/00 (2006.01) Wilmington, DE (US) CI2N 15/70 (2006.01) (52) U.S. C. (72) Inventors: Adriana Leonora Botes, Rosedale East CPC ................. CI2P5/007 (2013.01); C12N 15/70 (GB); Alex Van Eck Conradie, (2013.01) Eaglescliffe (GB) USPC ................... 435/167; 435/252.33: 435/252.3: 435/252.32; 435/252.34; 435/252.31; (21) Appl. No.: 14/452,201 435/254.3; 435/254.21: 435/254.23; 435/254.2: 435/254.11: 435/254.22 (22) Filed: Aug. 5, 2014 (57) ABSTRACT This document describes biochemical pathways for produc ing isoprene by forming two vinyl groups in a central precur Related U.S. Application Data sor produced from isobutyryl-CoA, 3-methyl-2-oxopen (60) Provisional application No. 61/862,401, filed on Aug. tanoate, or 4-methyl-2-oxopentanoate as well as recombinant 5, 2013. hosts for producing isoprene. Patent Application Publication Feb. 5, 2015 Sheet 1 of 19 US 2015/0037860 A1 FIGURE 1. O O CoA Acetyl-CoA --sca {YN 1 -sc.AO Acetoacetyl-CoA EC 2.3.1.9 Acetyl-CoA Acetyl-CoA t HO -->Cs.O OH Q CoA 3-Hydroxy 3-methylglutaryl-CoA gy 2.H a. CoA Co a 2NAD(P)* Jyu HO OH (R)-mevalonate t ATP c N N e d ADP O HO o–F–o OH (R)-5-phosphomevalonate t ATP c N s na No ADP JyuO HO O-P-O-P-OH OH OH (R)-5-Diphosphomevalonate t ATP c a g ADP PP CO2 N-1 -- P-O-P-OH HEos3.32 -- . -

SI Appendix Index 1
SI Appendix Index Calculating chemical attributes using EC-BLAST ................................................................................ 2 Chemical attributes in isomerase reactions ............................................................................................ 3 Bond changes …..................................................................................................................................... 3 Reaction centres …................................................................................................................................. 5 Substrates and products …..................................................................................................................... 6 Comparative analysis …........................................................................................................................ 7 Racemases and epimerases (EC 5.1) ….................................................................................................. 7 Intramolecular oxidoreductases (EC 5.3) …........................................................................................... 8 Intramolecular transferases (EC 5.4) ….................................................................................................. 9 Supporting references …....................................................................................................................... 10 Fig. S1. Overview …............................................................................................................................ -

6.2 Oleate Hydratase
Study Towards Carotenoid 1,2-Hydratase and Oleate Hydratase as Novel Biocatalysts Aida HISENI Study Towards Carotenoid 1,2-Hydratase and Oleate Hydratase as Novel Biocatalysts PROEFSCHRIFT ter verkrijging van de graad van doctor aan de Technische universiteit Delft, op gezag van de Rector Magnificus prof. ir. K.C.A.M Luyben, voorzitter van het College voor promoties, in het openbaar te verdedigen op dinsdag 22 april 2014 om 10:00 uur door Aida HISENI Diplom-Biologin, Heinrich-Heine-Universität Düsseldorf geboren te Doboj, Bosnië en Hercegovina. Dit proefschrift is goedgekeurd door de promotor: Prof. dr. I.W.C.E Arends Samenstelling promotiecommissie: Rector Magnificus voorzitter Prof. dr. I.W.C.E. Arends Technische Universiteit Delft, promotor Prof. dr. U. Hanefeld Technische Universiteit Delft Prof. dr. J.H. de Winde Universiteit Leiden Prof. dr. G. Muijzer Universiteit van Amsterdam Prof. dr. R. Wever Universiteit van Amsterdam Dr. L.G. Otten Technische Universiteit Delft Dr. P. Dominguez De Maria Sustainable Momentum Prof. dr. S. de Vries Technische Universiteit Delft, reservelid This project is financially supported by The Netherlands Ministry of Economic Affairs and the B-Basic partner organizations (http://www.b-basic.nl) through B-Basic, a public- private NWO-ACTS programme [Advanced Chemical Technologies for Sustainability (ACTS)]. ISBN Copyright © 2014 by Aida HISENI All rights reserved. No part of this publication may be reproduced or distributed in any form or by any means, or stored in a database or retrieval system, without any prior permission of the copyright owner. To my father Ismet Nukičić Table of Contents 1 General introduction ................................................................................................. -

Staphylococcus Aureus Adapts to the Host Nutritional Landscape to Overcome Tissue-Specific Branched-Chain Fatty Acid Requirement
Staphylococcus aureus adapts to the host nutritional landscape to overcome tissue-specific branched-chain fatty acid requirement Wei Ping Teoha, Xi Chena, Irina Laczkovicha, and Francis Alonzo IIIa,1 aDepartment of Microbiology and Immunology, Loyola University Chicago Stritch School of Medicine, Maywood, IL 60153 Edited by John E. Cronan, University of Illinois at Urbana–Champaign, Urbana, IL, and approved February 16, 2021 (received for review October 30, 2020) During infection, pathogenic microbes adapt to the nutritional is essential for the synthesis of saturated branched-chain fatty milieu of the host through metabolic reprogramming and nutrient acids (BCFAs), a major component of staphylococcal membrane scavenging. For the bacterial pathogen Staphylococcus aureus, vir- phospholipids (6). Like unsaturated fatty acids (UFAs), BCFAs ulence in diverse infection sites is driven by the ability to scavenge provide membrane fluidity to staphylococci, which neither syn- myriad host nutrients, including lipoic acid, a cofactor required for thesize UFAs nor encode a fatty acid (FA) desaturase that con- the function of several critical metabolic enzyme complexes. S. verts saturated fatty acids (SFAs) to unsaturated products (7). aureus shuttles lipoic acid between these enzyme complexes via Since a lack of membrane fluidity triggers cell death as a result of the amidotransferase, LipL. Here, we find that acquisition of lipoic lipid phase separation and protein segregation, bacterial survival acid, or its attachment via LipL to enzyme complexes required for hinges on the fine-tuning of membrane lipid composition to adapt the generation of acetyl-CoA and branched-chain fatty acids, is to fluctuating environments (8). essential for bacteremia, yet dispensable for skin infection in mice. -

Modelling of Substrate Access and Substrate Binding to Cephalosporin Acylases
www.nature.com/scientificreports OPEN Modelling of substrate access and substrate binding to cephalosporin acylases Received: 30 May 2019 Valerio Ferrario1, Mona Fischer1, Yushan Zhu2 & Jürgen Pleiss 1 Accepted: 14 August 2019 Semisynthetic cephalosporins are widely used antibiotics currently produced by diferent chemical Published: xx xx xxxx steps under harsh conditions, which results in a considerable amount of toxic waste. Biocatalytic synthesis by the cephalosporin acylase from Pseudomonas sp. strain N176 is a promising alternative. Despite intensive engineering of the enzyme, the catalytic activity is still too low for a commercially viable process. To identify the bottlenecks which limit the success of protein engineering eforts, a series of MD simulations was performed to study for two acylase variants (WT, M6) the access of the substrate cephalosporin C from the bulk to the active site and the stability of the enzyme-substrate complex. In both variants, cephalosporin C was binding to a non-productive substrate binding site (E86α, S369β, S460β) at the entrance to the binding pocket, preventing substrate access. A second non-productive binding site (G372β, W376β, L457β) was identifed within the binding pocket, which competes with the active site for substrate binding. Noteworthy, substrate binding to the protein surface followed a Langmuir model resulting in binding constants K = 7.4 and 9.2 mM for WT and M6, respectively, which were similar to the experimentally determined Michaelis constants KM = 11.0 and 8.1 mM, respectively. Semisynthetic cephalosporins are widely used antibiotics to protect against extended-spectrum β-lactamase producing pathogens. Cephalosporin antibiotics are usually synthesized from 7-amino cephalosporanic acid (7-ACA) which is obtained by the hydrolysis of cephalosporin C (CPC) available from fermentation (Fig. -

Chronic Pancreatitis.Pdf
CHRONIC PANCREATITIS: ELECTROPHILIC STRESS TEMPLATE Joan M Braganza DSc FRCP FRCPath DEVELOPMENT RATIONALISING GEOGRAPHY PETROCHEMICAL CONNECTION ACCOMMODATING GENE MUTATIONS MICRONUTRIENT THERAPY MAST CELL PATHOLOGY DISEASE PREVENTION CHRONIC PANCREATITIS: ELECTROPHILIC STRESS TEMPLATE By: Joan M Braganza DSc FRCP FRCPath University of Manchester Manchester, UK Published by: Pancreapedia: Exocrine Pancreas Knowledge Base In partnership with the University of Michigan Library Ann Arbor, MI 48109 and The American Pancreatic Association ISBN 978-1-60785-412-8 (e-book) This work is licensed under the Creative Commons Attribution-NonCommercial 4.0 International License. To view a copy of this license, visit https://creativecommons.org/licenses/by- nc/4.0/ or send a letter to Creative Commons, PO Box 1866, Mountain View, California, 94042, USA. Publishers Preface This monograph presents a comprehensive review of the work of Professor Joan Braganza into the pathogenesis and treatment of chronic pancreatitis as a disease caused by oxidative and particularly electrophilic stress with an emphasis on environmental toxicants as one factor which is part of the balance of damaging and protective cellular forces in the pancreas, liver and other participating organs. This is the second monograph of this type and follows “The Hamster Pancreas” written by Parviz Pour. Both books are published in open access eBook form under a Creative Commons license and are available on the Pancreapedia site. Pancreapedia will from time to time publish eBooks that are more substantive than a review article and where the author prepares the text and secures copyright approval for figures or tables published previously. Publishing can be done at no or low cost to the author. -
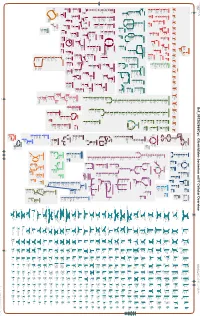
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Ron Caspi Peter Midford Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_001282665Cyc: Clostridiales bacterium mt11 Cellular Overview Connections between pathways are omitted for legibility. -

Under the Direction of Dr. David J. Dix, Dr
ABSTRACT GOETZ, AMBER KRISTINA. Toxicogenomic Study of Triazole Antifungal Modes of Action. (Under the direction of Dr. David J. Dix, Dr. Ernest Hodgson.) Common modes and mechanisms of hepatic and reproductive toxicity were characterized for a set of triazole antifungals. Wistar Han rats were fed myclobutanil, propiconazole, or triadimefon from gestation day 6 to postnatal day (PND) 120. Anogenital distance, body and organ weights, serum hormone levels, age at preputial separation, sperm morphology and motility, fertility and fecundity assays were selected to evaluate effects on development and adult reproductive function. All three triazoles increased anogenital distance, increased relative liver weights, induced hepatomegaly, increased absolute testis weights and serum testosterone levels. Myclobutanil and triadimefon impaired insemination and fertility, myclobutanil decreased pituitary weights, and triadimefon delayed puberty and decreased total serum thyroxine levels. The reproductive effects are consistent with disruption of testosterone homeostasis as a key event for triazole reproductive toxicity. To investigate common mechanisms of triazole toxicity, a toxicogenomic study was performed using liver and testis samples from PND92. Pathway and gene-level analysis of the liver highlighted biological processes affected by all three triazoles, including phase I-III; fatty acid, steroid, and xenobiotic metabolism; and lipid homeostasis. Triadimefon had a distinctive impact on sterol biosynthesis related genes in the liver. No common pathways were affected in the testis. To explore common mechanisms of action between in vivo and in vitro model systems, a series of comparative toxicogenomic studies were conducted on rat liver at multiple time points, rat and human primary hepatocytes, H295R cells, and PND92 liver and testis. Comparison between liver and rat hepatocytes showed consistent effect on fatty acid catabolism, sterol metabolism, and phase III transporters.