Modelling of Substrate Access and Substrate Binding to Cephalosporin Acylases
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Rhodococcus Erythropolis Oleate Hydratase: a New Member in the Oleate Hydratase Family Tree – Biochemical and Structural Studies
Technische Universität München Fakultät für Chemie Werner Siemens-Lehrstuhl für Synthetische Biotechnologie Enzymatic functionalization of bio based fatty acids and algae based triglycerides Jan Lorenzen Vollständiger Abdruck der von der Fakultät für Chemie der Technischen Universität München zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.) genehmigten Dissertation. Vorsitzender Prof. Dr. Tom Nilges Prüfer der Dissertation 1. Prof. Dr. Thomas Brück 2. Prof. Dr. Wolfgang Eisenreich 3. Prof. Dr. Uwe Bornscheuer (schriftliche Beurteilung) Prof. Dr. Thomas Fässler (mündliche Prüfung) Die Dissertation wurde am 26.09.2019 bei der Technischen Universität München eingereicht und durch die Fakultät für Chemie am 19.11.2019 angenommen. Abstracts I Abstracts Chapter I In the first chapter, the optimization of the SCCO2 extraction of microalgae derived biomass in an industrially relevant pilot plant that adheres to ATEX standards was reported for the first time. The SCCO2 extraction procedure of the lipid containing biomass of the microalgae strains Scenedesmus obliquus and Scenedesmus obtusiusculus was optimized with batch sizes of up to 1,3 kg of dried biomass. Under optimal conditions lipid recovery up to 92% w/w of the total lipids fraction could be achieved. Moreover, we addressed the purification of crude microalgae lipid extracts obtained by the SCCO2 extraction procedure. We could identify high amounts of hydrophobic contaminants (e.g. chlorophylls or carotenoids) in the crude lipid extract, which we could effectively remove by the application of the standard mineral adsorber bentonite. The obtained, clean microalgae oil can directly be further processed to lubricants, cosmetics or nutraceutical products. This is the first time that a combined SCCO2 extraction and purification process for algae lipids has been developed that could be integrated into existing industrial processes. -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota. -

(12) Patent Application Publication (10) Pub. No.: US 2015/0037860 A1 B0tes Et Al
US 2015 0037860A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2015/0037860 A1 B0tes et al. (43) Pub. Date: Feb. 5, 2015 (54) METHODS FOR BOSYNTHESIS OF Publication Classification ISOPRENE (51) Int. C. (71) Applicant: INVISTA North America S.ár.l., CI2P 5/00 (2006.01) Wilmington, DE (US) CI2N 15/70 (2006.01) (52) U.S. C. (72) Inventors: Adriana Leonora Botes, Rosedale East CPC ................. CI2P5/007 (2013.01); C12N 15/70 (GB); Alex Van Eck Conradie, (2013.01) Eaglescliffe (GB) USPC ................... 435/167; 435/252.33: 435/252.3: 435/252.32; 435/252.34; 435/252.31; (21) Appl. No.: 14/452,201 435/254.3; 435/254.21: 435/254.23; 435/254.2: 435/254.11: 435/254.22 (22) Filed: Aug. 5, 2014 (57) ABSTRACT This document describes biochemical pathways for produc ing isoprene by forming two vinyl groups in a central precur Related U.S. Application Data sor produced from isobutyryl-CoA, 3-methyl-2-oxopen (60) Provisional application No. 61/862,401, filed on Aug. tanoate, or 4-methyl-2-oxopentanoate as well as recombinant 5, 2013. hosts for producing isoprene. Patent Application Publication Feb. 5, 2015 Sheet 1 of 19 US 2015/0037860 A1 FIGURE 1. O O CoA Acetyl-CoA --sca {YN 1 -sc.AO Acetoacetyl-CoA EC 2.3.1.9 Acetyl-CoA Acetyl-CoA t HO -->Cs.O OH Q CoA 3-Hydroxy 3-methylglutaryl-CoA gy 2.H a. CoA Co a 2NAD(P)* Jyu HO OH (R)-mevalonate t ATP c N N e d ADP O HO o–F–o OH (R)-5-phosphomevalonate t ATP c N s na No ADP JyuO HO O-P-O-P-OH OH OH (R)-5-Diphosphomevalonate t ATP c a g ADP PP CO2 N-1 -- P-O-P-OH HEos3.32 -- . -

6.2 Oleate Hydratase
Study Towards Carotenoid 1,2-Hydratase and Oleate Hydratase as Novel Biocatalysts Aida HISENI Study Towards Carotenoid 1,2-Hydratase and Oleate Hydratase as Novel Biocatalysts PROEFSCHRIFT ter verkrijging van de graad van doctor aan de Technische universiteit Delft, op gezag van de Rector Magnificus prof. ir. K.C.A.M Luyben, voorzitter van het College voor promoties, in het openbaar te verdedigen op dinsdag 22 april 2014 om 10:00 uur door Aida HISENI Diplom-Biologin, Heinrich-Heine-Universität Düsseldorf geboren te Doboj, Bosnië en Hercegovina. Dit proefschrift is goedgekeurd door de promotor: Prof. dr. I.W.C.E Arends Samenstelling promotiecommissie: Rector Magnificus voorzitter Prof. dr. I.W.C.E. Arends Technische Universiteit Delft, promotor Prof. dr. U. Hanefeld Technische Universiteit Delft Prof. dr. J.H. de Winde Universiteit Leiden Prof. dr. G. Muijzer Universiteit van Amsterdam Prof. dr. R. Wever Universiteit van Amsterdam Dr. L.G. Otten Technische Universiteit Delft Dr. P. Dominguez De Maria Sustainable Momentum Prof. dr. S. de Vries Technische Universiteit Delft, reservelid This project is financially supported by The Netherlands Ministry of Economic Affairs and the B-Basic partner organizations (http://www.b-basic.nl) through B-Basic, a public- private NWO-ACTS programme [Advanced Chemical Technologies for Sustainability (ACTS)]. ISBN Copyright © 2014 by Aida HISENI All rights reserved. No part of this publication may be reproduced or distributed in any form or by any means, or stored in a database or retrieval system, without any prior permission of the copyright owner. To my father Ismet Nukičić Table of Contents 1 General introduction ................................................................................................. -

Staphylococcus Aureus Adapts to the Host Nutritional Landscape to Overcome Tissue-Specific Branched-Chain Fatty Acid Requirement
Staphylococcus aureus adapts to the host nutritional landscape to overcome tissue-specific branched-chain fatty acid requirement Wei Ping Teoha, Xi Chena, Irina Laczkovicha, and Francis Alonzo IIIa,1 aDepartment of Microbiology and Immunology, Loyola University Chicago Stritch School of Medicine, Maywood, IL 60153 Edited by John E. Cronan, University of Illinois at Urbana–Champaign, Urbana, IL, and approved February 16, 2021 (received for review October 30, 2020) During infection, pathogenic microbes adapt to the nutritional is essential for the synthesis of saturated branched-chain fatty milieu of the host through metabolic reprogramming and nutrient acids (BCFAs), a major component of staphylococcal membrane scavenging. For the bacterial pathogen Staphylococcus aureus, vir- phospholipids (6). Like unsaturated fatty acids (UFAs), BCFAs ulence in diverse infection sites is driven by the ability to scavenge provide membrane fluidity to staphylococci, which neither syn- myriad host nutrients, including lipoic acid, a cofactor required for thesize UFAs nor encode a fatty acid (FA) desaturase that con- the function of several critical metabolic enzyme complexes. S. verts saturated fatty acids (SFAs) to unsaturated products (7). aureus shuttles lipoic acid between these enzyme complexes via Since a lack of membrane fluidity triggers cell death as a result of the amidotransferase, LipL. Here, we find that acquisition of lipoic lipid phase separation and protein segregation, bacterial survival acid, or its attachment via LipL to enzyme complexes required for hinges on the fine-tuning of membrane lipid composition to adapt the generation of acetyl-CoA and branched-chain fatty acids, is to fluctuating environments (8). essential for bacteremia, yet dispensable for skin infection in mice. -
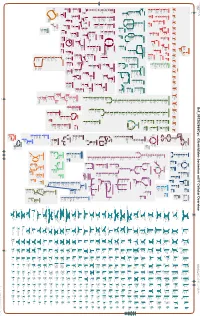
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Ron Caspi Peter Midford Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_001282665Cyc: Clostridiales bacterium mt11 Cellular Overview Connections between pathways are omitted for legibility. -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S. -

Catalysis Science & Technology
Catalysis Science & Technology Accepted Manuscript This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication. Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. We will replace this Accepted Manuscript with the edited and formatted Advance Article as soon as it is available. You can find more information about Accepted Manuscripts in the Information for Authors. Please note that technical editing may introduce minor changes to the text and/or graphics, which may alter content. The journal’s standard Terms & Conditions and the Ethical guidelines still apply. In no event shall the Royal Society of Chemistry be held responsible for any errors or omissions in this Accepted Manuscript or any consequences arising from the use of any information it contains. www.rsc.org/catalysis Page 1 of 34 Catalysis Science & Technology The selective addition of water Verena Resch a,b and Ulf Hanefeld* a a Gebouw voor Scheikunde, Biokatalyse, Afdeling Biotechnologie, Technische Universiteit Delft, Manuscript Julianalaan 136, 2628BL Delft, Nederland. bOrganische und Bioorganische Chemie, Institut für Chemie, Karl-Franzens-Universität Graz, Heinrichstrasse 28, 8010 Graz, Österreich. Abstract: Water is omnipresent and essential. Yet at the same time it is a rather unreactive Accepted molecule. The direct addition of water to C=C double bonds is therefore a challenge not answered convincingly. In this perspective we critically evaluate the selectivity and the applicability of the different catalytic approaches for water addition reactions, homogeneous, heterogeneous and bio- catalytic. -

POLSKIE TOWARZYSTWO BIOCHEMICZNE Postępy Biochemii
POLSKIE TOWARZYSTWO BIOCHEMICZNE Postępy Biochemii http://rcin.org.pl WSKAZÓWKI DLA AUTORÓW Kwartalnik „Postępy Biochemii” publikuje artykuły monograficzne omawiające wąskie tematy, oraz artykuły przeglądowe referujące szersze zagadnienia z biochemii i nauk pokrewnych. Artykuły pierwszego typu winny w sposób syntetyczny omawiać wybrany temat na podstawie możliwie pełnego piśmiennictwa z kilku ostatnich lat, a artykuły drugiego typu na podstawie piśmiennictwa z ostatnich dwu lat. Objętość takich artykułów nie powinna przekraczać 25 stron maszynopisu (nie licząc ilustracji i piśmiennictwa). Kwartalnik publikuje także artykuły typu minireviews, do 10 stron maszynopisu, z dziedziny zainteresowań autora, opracowane na podstawie najnow szego piśmiennictwa, wystarczającego dla zilustrowania problemu. Ponadto kwartalnik publikuje krótkie noty, do 5 stron maszynopisu, informujące o nowych, interesujących osiągnięciach biochemii i nauk pokrewnych, oraz noty przybliżające historię badań w zakresie różnych dziedzin biochemii. Przekazanie artykułu do Redakcji jest równoznaczne z oświadczeniem, że nadesłana praca nie była i nie będzie publikowana w innym czasopiśmie, jeżeli zostanie ogłoszona w „Postępach Biochemii”. Autorzy artykułu odpowiadają za prawidłowość i ścisłość podanych informacji. Autorów obowiązuje korekta autorska. Koszty zmian tekstu w korekcie (poza poprawieniem błędów drukarskich) ponoszą autorzy. Artykuły honoruje się według obowiązujących stawek. Autorzy otrzymują bezpłatnie 25 odbitek swego artykułu; zamówienia na dodatkowe odbitki (płatne) należy zgłosić pisemnie odsyłając pracę po korekcie autorskiej. Redakcja prosi autorów o przestrzeganie następujących wskazówek: Forma maszynopisu: maszynopis pracy i wszelkie załączniki należy nadsyłać w dwu egzem plarzach. Maszynopis powinien być napisany jednostronnie, z podwójną interlinią, z marginesem ok. 4 cm po lewej i ok. 1 cm po prawej stronie; nie może zawierać więcej niż 60 znaków w jednym wierszu nie więcej niż 30 wierszy na stronie zgodnie z Normą Polską. -

Title Characterization of the Linoleic Acid Δ9 Hydratase Catalyzing the First Step of Polyunsaturated Fatty Acid Saturation
Characterization of the linoleic acid Δ9 hydratase catalyzing Title the first step of polyunsaturated fatty acid saturation metabolism in Lactobacillus plantarum AKU 1009a. Takeuchi, Michiki; Kishino, Shigenobu; Hirata, Akiko; Park, Author(s) Si-Bum; Kitamura, Nahoko; Ogawa, Jun Journal of bioscience and bioengineering (2014), 119(6): 636- Citation 641 Issue Date 2014-12-01 URL http://hdl.handle.net/2433/198741 © 2014 The Society for Biotechnology, Japan. Licensed under the Creative Commons Attribution-NonCommercial- NoDerivatives 4.0 International http://creativecommons.org/licenses/by-nc-nd/4.0/. NOTICE: this is the author's version of a work that was accepted for publication in <Journal of Bioscience and Bioengineering>. Changes resulting from the publishing process, such as peer review, editing, corrections, structural formatting, and other quality control mechanisms may not be reflected in this Right document. Changes may have been made to this work since it was submitted for publication. A definitive version was subsequently published in [Journal of Bioscience and Bioengineering, Volume 119, Issue 6, Pages 636‒641] doi:10.1016/j.jbiosc.2014.10.022.; 許諾条件により本文ファ イルは2015-12-01に公開.; この論文は出版社版でありませ ん。引用の際には出版社版をご確認ご利用ください。; This is not the published version. Please cite only the published version. Type Journal Article Textversion author Kyoto University 1 Characterization of the linoleic acid Δ9 hydratase catalyzing the first step of 2 polyunsaturated fatty acid saturation metabolism in Lactobacillus plantarum AKU 3 1009a 4 Running -

On the Michael Addition of Water to C = C Bonds
ON THE MICHAEL ADDITION OF WATER TO C = C BONDS ON THE MICHAEL ADDITION OF WATER TO C = C BONDS Proefschrift ter verkrijging van de graad van doctor aan de Technische Universiteit Delft, op gezag van de RectorMagnificus Prof. ir. K. C. A. M. Luyben, voorzitter van het College voor Promoties, in het openbaar te verdedigen op dinsdag 01 September 2015 om 10:00 uur door Bishuang CHEN Master in Organic Chemistry, Xiamen University, Xiamen, China geboren te Putian, Fujian Province, China Dit proefschrift is goedgekeurd door de promotoren: Prof. dr. Ulf Hanefeld Samenstelling promotiecommissie: Rector Magnificus, voorzitter Prof. dr. Ulf Hanefeld, Technische Universiteit Delft, promotor Prof. dr. W.R. Hagen, Technische Universiteit Delft Dr. ir. F. Hollmann Technische Universiteit Delft Prof. dr. W.J.H. van Berkel Wageningen University Prof. dr. J.G. Roelfes University of Groningen Prof. dr. H. Gröger Bielefeld University, Duitsland Dr. V.A. Resch University of Graz, Graz Prof. dr. I.W.C.E. Arends Technische Universiteit Delft, reserved water, Michael hydratase, ene-reductase, Rhodococcus, Keywords: -hydroxy carbonyl compound Printed by: Ipskamp Drukkers biocatalysis, lipase, β Cover by: Huayang Cai ISBN: 978-94-6259-773-0 Copyright © 2015 Bishuang CHEN An electronic version of this dissertation is available at http://repository.tudelft.nl/ All rights reserved. No parts of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photo-copying, recording, or otherwise, without the prior written permission of the author. To my parents Contents 1 Stereochemistry of enzymatic water addition to C=C bonds .................. -

(12) Patent Application Publication (10) Pub. No.: US 2012/0266329 A1 Mathur Et Al
US 2012026.6329A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2012/0266329 A1 Mathur et al. (43) Pub. Date: Oct. 18, 2012 (54) NUCLEICACIDS AND PROTEINS AND CI2N 9/10 (2006.01) METHODS FOR MAKING AND USING THEMI CI2N 9/24 (2006.01) CI2N 9/02 (2006.01) (75) Inventors: Eric J. Mathur, Carlsbad, CA CI2N 9/06 (2006.01) (US); Cathy Chang, San Marcos, CI2P 2L/02 (2006.01) CA (US) CI2O I/04 (2006.01) CI2N 9/96 (2006.01) (73) Assignee: BP Corporation North America CI2N 5/82 (2006.01) Inc., Houston, TX (US) CI2N 15/53 (2006.01) CI2N IS/54 (2006.01) CI2N 15/57 2006.O1 (22) Filed: Feb. 20, 2012 CI2N IS/60 308: Related U.S. Application Data EN f :08: (62) Division of application No. 1 1/817,403, filed on May AOIH 5/00 (2006.01) 7, 2008, now Pat. No. 8,119,385, filed as application AOIH 5/10 (2006.01) No. PCT/US2006/007642 on Mar. 3, 2006. C07K I4/00 (2006.01) CI2N IS/II (2006.01) (60) Provisional application No. 60/658,984, filed on Mar. AOIH I/06 (2006.01) 4, 2005. CI2N 15/63 (2006.01) Publication Classification (52) U.S. Cl. ................... 800/293; 435/320.1; 435/252.3: 435/325; 435/254.11: 435/254.2:435/348; (51) Int. Cl. 435/419; 435/195; 435/196; 435/198: 435/233; CI2N 15/52 (2006.01) 435/201:435/232; 435/208; 435/227; 435/193; CI2N 15/85 (2006.01) 435/200; 435/189: 435/191: 435/69.1; 435/34; CI2N 5/86 (2006.01) 435/188:536/23.2; 435/468; 800/298; 800/320; CI2N 15/867 (2006.01) 800/317.2: 800/317.4: 800/320.3: 800/306; CI2N 5/864 (2006.01) 800/312 800/320.2: 800/317.3; 800/322; CI2N 5/8 (2006.01) 800/320.1; 530/350, 536/23.1: 800/278; 800/294 CI2N I/2 (2006.01) CI2N 5/10 (2006.01) (57) ABSTRACT CI2N L/15 (2006.01) CI2N I/19 (2006.01) The invention provides polypeptides, including enzymes, CI2N 9/14 (2006.01) structural proteins and binding proteins, polynucleotides CI2N 9/16 (2006.01) encoding these polypeptides, and methods of making and CI2N 9/20 (2006.01) using these polynucleotides and polypeptides.