Physics Reports a Perspective on Conventional High
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Densifying Metal Hydrides with High Temperature and Pressure
3,784,682 United States Patent Office Patented Jan. 8, 1974 feet the true density. That is, by this method only theo- 3,784,682 retical or near theoretical densities can be obtained by DENSIFYING METAL HYDRIDES WITH HIGH making the material quite free from porosity (p. 354). TEMPERATURE AND PRESSURE The true density remains the same. Leonard M. NiebylsM, Birmingham, Mich., assignor to Ethyl Corporation, Richmond, Va. SUMMARY OF THE INVENTION No Drawing. Continuation-in-part of abandoned applica- tion Ser. No. 392,370, Aug. 24, 1964. This application The process of this invention provides a practical Apr. 9,1968, Ser. No. 721,135 method of increasing the true density of hydrides of Int. CI. COlb 6/00, 6/06 metals of Groups II-A, II-B, III-A and III-B of the U.S. CI. 423—645 8 Claims Periodic Table. More specifically, true densities of said 10 metal hydrides may be substantially increased by subject- ing a hydride to superatmospheric pressures at or above ABSTRACT OF THE DISCLOSURE fusion temperatures. When beryllium hydride is subjected A method of increasing the density of a hydride of a to this process, a material having a density of at least metal of Groups II-A, II-B, III-A and III-B of the 0.69 g./cc. is obtained. It may or may not be crystalline. Periodic Table which comprises subjecting a hydride to 15 a pressure of from about 50,000 p.s.i. to about 900,000 DESCRIPTION OF THE PREFERRED p.s.i. at or above the fusion temperature of the hydride; EMBODIMENT i.e., between about 65° C. -

Recent Studies in Superconductivity at Extreme Pressures
Recent Studies in Superconductivity at Extreme Pressures James S. Schilling and James J. Hamlin Department of Physics, Washington University CB 1105, One Brookings Dr, St. Louis, MO 63130, USA October 12, 2007 Abstract Studies of the effect of high pressure on superconductivity began in 1925 with the seminal work of Sizoo and Onnes on Sn to 0.03 GPa and have continued up to the present day to pressures in the 200 - 300 GPa range. Such enormous pressures cause profound changes in all condensed matter properties, including superconductivity. In high pressure experiments metallic elements, Tc values have been elevated to temperatures as high as 20 K for Y at 115 GPa and 25 K for Ca at 160 GPa. These pressures are sufficient to turn many insulators into metals and magnetics into superconductors. The changes will be particularly dramatic when the pressure is sufficient to break up one or more atomic shells. Recent results in superconductivity to Mbar pressures wll be discussed which exemplify the progress made in this field over the past 82 years. 1 Superconductivity is a macroscopic quantum phenomenon which was discovered by G. J. Holst and H. K. Onnes [1] in Leiden in 1911, but not clearly understood untilBardeen,Cooper,andSchrieffer (BCS) [2] formulated their microscopic theory in 1957, exactly half a century ago. In the opinion of one of the present authors (JSS), had superconductivity not first been demonstrated in experiment, no theorist would have ever predicted it: who could imagine that two electrons, in spite of their Coulomb repulsion, might experience a net attractive interaction binding them together to form a bose particle? In a lecture in 1922 in honor of H. -

14Th International Ceramics Congress June 4-8/2018 Forum on New Materials June 10-14/2018
Astract Deadline • October 15, 2017 Perugia - Italy • June 4-14/2018 14th International Ceramics Congress June 4-8/2018 8th Forum on New Materials June 10-14/2018 www.cimtec-congress.org Benedetto Bonfigli, Porta Marzia in Perugia, XV sec. Invitation to Attend! CIMTEC 2018 - 14th International Conference on Modern Materials and Technologies - will be held in Peru- gia, Italy, June 4 to 14, 2018. CIMTEC 2018 will consist of the 14th International Ceramics Congress (June 4-8) and of the 8th Forum on New Materials (June 10-14), each of them including a number of Symposia, Special Sessions, and Conferences. As a major longstanding event for the international materials community, CIMTEC will again gather together a large and qualified audience of materials scientists, physicists, chemists and engineers and of experts of a wide range of the most demanding application areas of modern materials, from the molecular and nanoscales to large complex integrated systems. The National Research Council of Italy (CNR), the Italian National Agency for New Technology, Energy and the Environment (ENEA) will act as major endorsers of CIMTEC 2018 together with the World Academy of Ceramics (WAC), The International Ceramic Federation (ICF) and the International Union of the Materials Research Societies (IUMRS). The Chair, Co-Chairs and CIMTEC 2018 Committees invite you to foster the progress in the field by contrib- uting with your expertise to what promises to be a very comprehensive and exciting meeting, and to enjoy the immense unique artistic heritage and wonderful landscape of Umbria district. Pietro Vincenzini Co-Chairs CIMTEC 2018 General Chair CIMTEC Conferences Gary Messing National Research Council, Italy Penn State University, USA World Academy of Ceramics Takashi Goto Tohoku University, Japan International Ceramic Federation Robert P.H. -

Underpinning of Soviet Industrial Paradigms
Science and Social Policy: Underpinning of Soviet Industrial Paradigms by Chokan Laumulin Supervised by Professor Peter Nolan Centre of Development Studies Department of Politics and International Studies Darwin College This dissertation is submitted for the degree of Doctor of Philosophy May 2019 Preface This dissertation is the result of my own work and includes nothing which is the outcome of work done in collaboration except as declared in the Preface and specified in the text. It is not substantially the same as any that I have submitted, or, is being concurrently submitted for a degree or diploma or other qualification at the University of Cambridge or any other University or similar institution except as declared in the Preface and specified in the text. I further state that no substantial part of my dissertation has already been submitted, or, is being concurrently submitted for any such degree, diploma or other qualification at the University of Cambridge or any other University or similar institution except as declared in the Preface and specified in the text It does not exceed the prescribed word limit for the relevant Degree Committee. 2 Chokan Laumulin, Darwin College, Centre of Development Studies A PhD thesis Science and Social Policy: Underpinning of Soviet Industrial Development Paradigms Supervised by Professor Peter Nolan. Abstract. Soviet policy-makers, in order to aid and abet industrialisation, seem to have chosen science as an agent for development. Soviet science, mainly through the Academy of Sciences of the USSR, was driving the Soviet industrial development and a key element of the preparation of human capital through social programmes and politechnisation of the society. -

Inorganic Seminar Abstracts
C 1 « « « • .... * . i - : \ ! -M. • ~ . • ' •» »» IB .< L I B RA FLY OF THE. UN IVERSITY Of 1LLI NOIS 546 1^52-53 Return this book on or before the Latest Date stamped below. University of Illinois Library «r L161— H41 Digitized by the Internet Archive in 2012 with funding from University of Illinois Urbana-Champaign http://archive.org/details/inorganicsemi195253univ INORGANIC SEMINARS 1952 - 1953 TABLE OF CONTENTS 1952 - 1953 Page COMPOUNDS CONTAINING THE SILICON-SULFUR LINKAGE 1 Stanley Kirschner ANALYTICAL PROCEDURES USING ACETIC ACID AS A SOLVENT 5 Donald H . Wilkins THE SOLVENT PHOSPHORYL CHLORIDE, POCl 3 12 S.J. Gill METHODS FOR PREPARATION OF PURE SILICON 17 Alex Beresniewicz IMIDODISULFINAMIDE 21 G.R. Johnston FORCE CONSTANTS IN POLYATOMIC MOLECILES 28 Donn D. Darsow METATHESIS IN LIQUID ARSENIC TRICHLORIDE 32 Harold H. Matsuguma THE RHENI DE OXIDATION STATE 40 Robert L. Rebertus HALOGEN CATIONS 45 L.H. Diamond REACTIONS OF THE NITROSYL ION 50 M.K. Snyder THE OCCURRENCE OF MAXIMUM OXIDATION STATES AMONG THE FLUOROCOMPLEXES OF THE FIRST TRANSITION SERIES 56 D.H. Busch POLY- and METAPHOSPHATES 62 V.D. Aftandilian PRODUCTION OF SILICON CHLORIDES BY ELECTRICAL DISCHARGE AND HIGH TEMPERATURE TECHNIQUES 67 VI. £, Cooley FLUORINE CONTAINING OXYHALIDES OF SULFUR 72 E.H. Grahn PREPARATION AND PROPERTIES OF URANYL CARBONATES 76 Richard *• Rowe THE NATURE OF IODINE SOLUTIONS 80 Ervin c olton SOME REACTIONS OF OZONE 84 Barbara H. Weil ' HYDRAZINE BY ELECTROLYSIS IN LIQUID AMMONIA 89 Robert N. Hammer NAPHTHAZARIN COMPLEXES OF THORIUM AND RARE EARTH METAL IONS 93 Melvin Tecotzky THESIS REPORT 97 Perry Kippur ION-PAIR FORMATION IN ACETIC ACID 101 M.M. -

(12) United States Patent (10) Patent No.: US 7,732,534 B2 Luo Et Al
US007732534B2 (12) United States Patent (10) Patent No.: US 7,732,534 B2 Luo et al. (45) Date of Patent: Jun. 8, 2010 (54) POLYMERS FUNCTIONALIZED WITH 6,172,160 B1 1/2001 Nakamura et al. NITRO COMPOUNDS 6, 194505 B1 2/2001 Sone et al. .................. 524/432 6, 197,888 B1 3/2001 Luo ........................... 525,247 (75) Inventors: Steven Luo, Copley, OH (US); Ryuji Nakagawa, Tokyo (JP) 6.255.416 B1 7/2001 Sone et al. .................. 526,153 6.271,315 B1* 8/2001 Kiessling et al. ......... 525,326.1 (73) Assignee: Bridgestone Corporation (JP) 6,291,591 B1 9/2001 Luo ........................... 525, 191 6,303,692 B1 10/2001 Luo ........................... 525, 191 (*) Notice: Subject to any disclaimer, the term of this 6,699,813 B2 3/2004 Luo et al. ................... 502,119 patent is extended or adjusted under 35 6,759,497 B2 7/2004 Grun et al. U.S.C. 154(b) by 526 days. 6,838,526 B1 1/2005 Sone et al. (21) Appl. No.: 11/710,845 6,897,270 B2 5/2005 Ozawa et al. ................. 526,88 6,977.281 B1* 12/2005 Ozawa et al. ............... 525/377 (22) Filed: Feb. 26, 2007 6,992,147 B1 1/2006 Ozawa et al. ............... 525,342 7,008,899 B2 3/2006 Luo et al. ................... 502,131 Prior Publication Data (65) 7,094,849 B2 8/2006 Luo et al. ................... 526, 164 US 2008/OO51552 A1 Feb. 28, 2008 7,351,776 B2 4/2008 Tartamella et al. Related U.S. Application Data 2004/O147694 A1 7/2004 Sone et al. -

Ternary Superconducting Cophosphorus Hydrides Stabilized Via Lithium
www.nature.com/npjcompumats ARTICLE OPEN Ternary superconducting cophosphorus hydrides stabilized via lithium Ziji Shao1, Defang Duan 1*, Yanbin Ma2, Hongyu Yu1, Hao Song1, Hui Xie1,DaLi 1, Fubo Tian1, Bingbing Liu1 and Tian Cui1* Inspired by the diverse properties of sulfur hydrides and phosphorus hydrides, we combine first-principles calculations with structure prediction to search for stable structures of Li−P−H ternary compounds at high pressures with the aim of finding novel superconductors. It is found that phosphorus hydrides can be stabilized under pressure via additional doped lithium. Four stable stoichiometries LiPH3, LiPH4, LiPH6, and LiPH7 are uncovered in the pressure range of 100–300 GPa. Notably, we find an atomic LiPH6 with Pm3 symmetry which is predicted to be a potential high-temperature superconductor with a Tc value of 150–167 K at 200 GPa and the Tc decreases upon compression. All the predicted stable ternary hydrides contain the P–H covalent frameworks with ionic lithium staying beside, but not for Pm3-LiPH6. We proposed a possible synthesis route for ternary lithium phosphorus hydrides: LiP + H2 → LiPHn, which could provide helpful and clear guidance to further experimental studies. Our work may provide some advice on further investigations on ternary superconductive hydrides at high pressure. npj Computational Materials (2019) 5:104 ; https://doi.org/10.1038/s41524-019-0244-6 1234567890():,; INTRODUCTION Experimentally, the PH3 is found to be gradually decomposed Given the metallization and potential room temperature super- into P2H4,P4H6, and then further into elemental phases above 22,23 conductivity of solid hydrogen under high pressure have been 35 GPa at room temperature. -
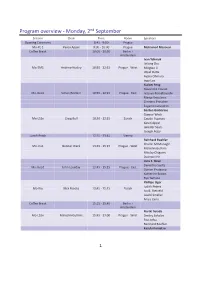
Program Overview ‐ Monday, 2ⁿd September
Program overview ‐ Monday, 2ⁿd September Session Chair Time Room Speakers Opening Ceremony 8:45 ‐ 9:00 Prague Mo‐PL 1 Karen Appel 9:00 ‐ 10:00 Prague Mohamed Mezouar Coffee Break 10:00 ‐ 10:30 Berlin + Amsterdam Ivan Yahniuk Jinlong Zhu Mo‐EM1 Andrew Huxley 10:30 ‐ 12:15 Prague ‐ West Mingtao Li Utpal Dutta Ayako Ohmura Ingo Loa Xialoei Feng Alexandre Courac Mo‐Geo1 Simon Redfern 10:30 ‐ 12:15 Prague ‐ East Tetsuya Komabayashi Marija Krstulovic Clemens Prescher Eugenio Calandrini Gaston Garbarino Gunnar Weck Mo‐LS1x Craig Bull 10:30 ‐ 12:15 Zürich Catalin Popescu Karen Appel Jennifer Sears Joseph Boby Lunch Break 12:15 ‐ 13:45 Vienna Reinhard Boehler Charlie McMonagle Mo‐Ins1 Gunnar Weck 13:45 ‐ 15:15 Prague ‐ West Malcolm Guthrie Nikolay Chigarev Duanwei He Livia E. Bove Demetrio Scelta Mo‐Geo2 John Loveday 13:45 ‐ 15:15 Prague ‐ East Ciprian Pruteanu Katherine Brown Ryo Yamane Phillipe Oger Judith Peters Mo‐Bio Nick Brooks 13:45 ‐ 15:15 Zürich Ivo B. Rietveld László Smeller Anais Cario Coffee Break 15:15 ‐ 15:45 Berlin + Amsterdam Noriki Terada Mo‐LS2n Malcolm Guthrie 15:45 ‐ 17:00 Prague ‐ West Dmitry Sokolov Pau Jorba Reinhard Boehler Kazuki Komatsu 1 Program overview ‐ Monday, 2ⁿd September Mo‐Geo3 Livia E. Bove 15:45 ‐ 17:00 Prague ‐ East Samuele Fanetti Dane Sterbentz Sooheyong Lee Kateřina Štulíková Mo‐Food Jorge Saraiva 15:45 ‐ 17:00 Zürich Concepción P. Lamela Izabela Porębska Milan Houška Poster Session 1 17:00 ‐ 18:45 Berlin 8:45 ‐ 9:00 Opening Ceremony Room Prague 9:00 ‐ 10:00 Plenary Lecture 1 Room Prague Chair Karen Appel -

Thesis Reference
Thesis Experimental and theoretical studies of boron and hydrogen containing compounds in relation to potential hydrogen storage and ionic conduction applications SHARMA, Manish Abstract This thesis deals with the fundamental studies of some materials containing boron-hydrogen bonds which can potentially be used either as the hydrogen storage materials (M(BH4)2, M=Alkaline earth metal), as the solid electrolytes for batteries (Na2B12H12) or as reducing agents for CO2 (Mg(BH4)2). First part of thesis deals with borohydrides (BH4-). Synthesis and characterization of halide-free Sr(BH4)2, Ba(BH4)2 and Eu(BH4)2 is reported. Crystallographic study of these compounds helped in identifying several new phases and a new species metal borohydride hydride (M2(BH4)H3). In depth study of B-H bond breaking is reported via isotope exchange reaction in Ca(BH4)2.A practical example of borohydride as reducing agent is reported by showing the reduction of CO2 with gamma-Mg(BH4)2. The second part of the thesis focuses on closoboranes derived from the B12H122- ion. Compounds of this family have recently attracted great interest as solid ionic conductors for Li and Na ions.Results of DFT calculations on isolated B12H122- anions and halogen (F, Cl or Br) substituted anions were analysed in detail. Synthesis of Na2B12(SCN)H11 is [...] Reference SHARMA, Manish. Experimental and theoretical studies of boron and hydrogen containing compounds in relation to potential hydrogen storage and ionic conduction applications. Thèse de doctorat : Univ. Genève, 2017, no. Sc. 5101 DOI : 10.13097/archive-ouverte/unige:96376 URN : urn:nbn:ch:unige-963769 Available at: http://archive-ouverte.unige.ch/unige:96376 Disclaimer: layout of this document may differ from the published version. -

55 Th EHPRG Guide.Pdf
www.ehprg2017.org 55th European High Pressure Research Group Meeting: High Pressure Science and Technology PROGRAMME 1 55th EHPRG Poznań, 3-8 September 2017 Programme Sunday, Monday, Tuesday, Wednesday, Thursday, Friday, Programme Overview Time schedule Time schedule 3 September 2017 4 September 2017 5 September 2017 6 September 2017 7 September 2017 8 September 2017 8.00-8.30 Registration 8.00-8.30 Registration Registration Special Session: Morasko Registration 8.30-9.00 Opening Ceremony 8.45 8.30-9.00 Meteorite Reserve 9.00-9.30 PL2 - Natalia 9.00-9.30 PL1 - Ho-Kwang Mao PL3 - Chris Michiels Mirosław Makohonienko PL5 - Przemysław Dera Dubrovinskaia Room 2.64 Room 2.64 Start in Room 2.64 Room 2.64 9.30-10.00 Room 2.64 9.30-10.00 10.00-10.30 Coffee Break Coffee Break Coffee Break Coffee Break Coffee Break 10.00-10.30 10.30-11.00 S 1 S 2 S 3 S 6 S 9 S 10 S 11 S 12 S 17 S 18 S 20 S 22 S 23 S 24 S 26 S 27 S 28 10.30-11.00 11.00-11.30 2.64 2.61 3.65 2.62 2.62 2.61 3.65 2.64 3.65 2.62 2.64 2.62 3.65 2.64 2.62 3.65 2.64 11.00-11.30 11.30-12.00 11.30-12.00 12.00-12.30 12.00-12.30 12.30-13.00 Lunch Break/Special Closing Ceremony 12.40 12.30-13.00 Lunch Break EHPRG General Session: “Women Under 13.00-13.30 Lunch Break Assembly 13.00-13.30 Pressure gathering” Room 2.64 Lunch 13.30-14.00 EHPRG Group Photo Room 2.57 13.30-14.00 14.00-14.30 S 5 S 4 S 7 S 8 S 13 S 14 S 15 S 16 PL4 - Elena Boldyreva 14.00-14.30 Lunch Break 14.30-15.00 2.64 2.61 3.65 2.62 3.65 2.61 2.64 2.62 Room 2.64 14.30-15.00 15.00-15.30 15.00-15.30 15.30-16.00 Poster Session 2 15.30-16.00 -

Download This Article PDF Format
RSC Advances PAPER View Article Online View Journal | View Issue Pressure-induced metallization in MoSe2 under different pressure conditions† Cite this: RSC Adv.,2019,9,5794 Linfei Yang,ab Lidong Dai, *a Heping Li,a Haiying Hu,a Kaixiang Liu,ab Chang Pu,ab Meiling Hongab and Pengfei Liuc In this study, the vibrational and electrical transport properties of molybdenum diselenide were investigated under both non-hydrostatic and hydrostatic conditions up to 40.2 GPa using the diamond anvil cell in conjunction with Raman spectroscopy, electrical conductivity, high-resolution transmission electron microscopy, atomic force microscopy, and first-principles theoretical calculations. The results obtained indicated that the semiconductor-to-metal electronic phase transition of MoSe2 can be extrapolated by some characteristic parameters including abrupt changes in the full width at half maximum of Raman modes, electrical conductivity and calculated bandgap. Under the non-hydrostatic condition, metallization occurred at 26.1 GPa and it was irreversible. However, reversible metallization occurred at 29.4 GPa under the hydrostatic condition. In addition, the pressure-induced metallization reversibility Creative Commons Attribution 3.0 Unported Licence. Received 16th November 2018 of MoSe can be revealed by high-resolution transmission electron and atomic force microscopy of the Accepted 4th February 2019 2 recovered samples under different hydrostatic conditions. This discrepancy in the metallization DOI: 10.1039/c8ra09441a phenomenon of MoSe2 in different hydrostatic environments was attributed to the mitigated interlayer rsc.li/rsc-advances van der Waals coupling and shear stress caused by the insertion of pressure medium into the layers. Introduction understanding the crystalline structure evolution and electrical properties of AB2-type layered materials, and also promotes its ¼ ¼ The AB2-type (A Mo, W; B S, Se, Te) transition-metal industrial exploitation in electronic devices. -

Pushing Towards Room-Temperature Superconductivity
VIEWPOINT Pushing Towards Room-Temperature Superconductivity Two independent studies report superconductivity at record high temperatures in hydrogen-rich materials under extreme pressure. by Eva Zurek∗ groups, the first led by Russell Hemley at the George Wash- ington University in Washington, DC [2], and the second by uperconductivity, the ability of a material to conduct Mikhail Eremets at the Max Planck Institute for Chemistry, electricity without any resistance, was first observed Germany [3], have now reported experiments indicating that in 1911 in solid mercury below a critical temperature a hydride of lanthanum compressed to 170–185 GPa has a Tc (Tc) of 4.2 K. Ever since, countless scientists have been of 250–260 K [2, 3]. The results bode well for the search for Ssearching for a material whose Tc exceeds room tempera- room-temperature superconductors—the reported materials ture. For a long time this holy grail seemed unattainable—a could already work without the need for cooling on an aver- linear extrapolation of research progress from 1911 to 1970 age winter night in the Arctic! suggested that Tc would reach room temperature around In 1968, physicist Neil Ashcroft predicted that metallic the year 2840! The discovery of high-temperature super- hydrogen should have all of the properties required to be conductivity in copper oxides raised Tc above liquid helium a high-temperature superconductor according to Bardeen- temperature. Since 1994, one of the copper oxides has held Cooper-Schrieffer (BCS) theory [4]. Unfortunately, metalliz- the record for the highest Tc (133 K at atmospheric pressure ing hydrogen in static compression experiments turned out and 164 K under high pressure).