Molecular Cloning and Functional Characterization of Benzophenone Synthases from Centaurium Erythraea and Swertia Chirata (Gentianaceae)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Flowering Plants Eudicots Apiales, Gentianales (Except Rubiaceae)
Edited by K. Kubitzki Volume XV Flowering Plants Eudicots Apiales, Gentianales (except Rubiaceae) Joachim W. Kadereit · Volker Bittrich (Eds.) THE FAMILIES AND GENERA OF VASCULAR PLANTS Edited by K. Kubitzki For further volumes see list at the end of the book and: http://www.springer.com/series/1306 The Families and Genera of Vascular Plants Edited by K. Kubitzki Flowering Plants Á Eudicots XV Apiales, Gentianales (except Rubiaceae) Volume Editors: Joachim W. Kadereit • Volker Bittrich With 85 Figures Editors Joachim W. Kadereit Volker Bittrich Johannes Gutenberg Campinas Universita¨t Mainz Brazil Mainz Germany Series Editor Prof. Dr. Klaus Kubitzki Universita¨t Hamburg Biozentrum Klein-Flottbek und Botanischer Garten 22609 Hamburg Germany The Families and Genera of Vascular Plants ISBN 978-3-319-93604-8 ISBN 978-3-319-93605-5 (eBook) https://doi.org/10.1007/978-3-319-93605-5 Library of Congress Control Number: 2018961008 # Springer International Publishing AG, part of Springer Nature 2018 This work is subject to copyright. All rights are reserved by the Publisher, whether the whole or part of the material is concerned, specifically the rights of translation, reprinting, reuse of illustrations, recitation, broadcasting, reproduction on microfilms or in any other physical way, and transmission or information storage and retrieval, electronic adaptation, computer software, or by similar or dissimilar methodology now known or hereafter developed. The use of general descriptive names, registered names, trademarks, service marks, etc. in this publication does not imply, even in the absence of a specific statement, that such names are exempt from the relevant protective laws and regulations and therefore free for general use. -

FLORA from FĂRĂGĂU AREA (MUREŞ COUNTY) AS POTENTIAL SOURCE of MEDICINAL PLANTS Silvia OROIAN1*, Mihaela SĂMĂRGHIŢAN2
ISSN: 2601 – 6141, ISSN-L: 2601 – 6141 Acta Biologica Marisiensis 2018, 1(1): 60-70 ORIGINAL PAPER FLORA FROM FĂRĂGĂU AREA (MUREŞ COUNTY) AS POTENTIAL SOURCE OF MEDICINAL PLANTS Silvia OROIAN1*, Mihaela SĂMĂRGHIŢAN2 1Department of Pharmaceutical Botany, University of Medicine and Pharmacy of Tîrgu Mureş, Romania 2Mureş County Museum, Department of Natural Sciences, Tîrgu Mureş, Romania *Correspondence: Silvia OROIAN [email protected] Received: 2 July 2018; Accepted: 9 July 2018; Published: 15 July 2018 Abstract The aim of this study was to identify a potential source of medicinal plant from Transylvanian Plain. Also, the paper provides information about the hayfields floral richness, a great scientific value for Romania and Europe. The study of the flora was carried out in several stages: 2005-2008, 2013, 2017-2018. In the studied area, 397 taxa were identified, distributed in 82 families with therapeutic potential, represented by 164 medical taxa, 37 of them being in the European Pharmacopoeia 8.5. The study reveals that most plants contain: volatile oils (13.41%), tannins (12.19%), flavonoids (9.75%), mucilages (8.53%) etc. This plants can be used in the treatment of various human disorders: disorders of the digestive system, respiratory system, skin disorders, muscular and skeletal systems, genitourinary system, in gynaecological disorders, cardiovascular, and central nervous sistem disorders. In the study plants protected by law at European and national level were identified: Echium maculatum, Cephalaria radiata, Crambe tataria, Narcissus poeticus ssp. radiiflorus, Salvia nutans, Iris aphylla, Orchis morio, Orchis tridentata, Adonis vernalis, Dictamnus albus, Hammarbya paludosa etc. Keywords: Fărăgău, medicinal plants, human disease, Mureş County 1. -

Recovery Plan for Tyoj5llllt . I-Bland Plants
Recovery Plan for tYOJ5llllt. i-bland Plants RECOVERY PLAN FOR MULTI-ISLAND PLANTS Published by U.S. Fish and Wildlife Service Portland, Oregon Approved: Date: / / As the Nation’s principal conservation agency, the Department of the Interior has responsibility for most ofour nationally owned public lands and natural resources. This includes fostering the wisest use ofour land and water resources, protecting our fish and wildlife, preserving the environmental and cultural values ofour national parks and historical places, and providing for the enjoyment of life through outdoor recreation. The Department assesses our energy and mineral resources and works to assure that their development is in the best interests ofall our people. The Department also has a major responsibility for American Indian reservation communities and for people who live in island Territories under U.S. administration. DISCLAIMER PAGE Recovery plans delineate reasonable actions that are believed to be required to recover and/or protect listed species. Plans are published by the U.S. Fish and Wildlife Service, sometimes prepared with the assistance ofrecovery teams, contractors, State agencies, and others. Objectives will be attained and any necessary funds made available subject to budgetary and other constraints affecting the parties involved, as well as the need to address other priorities. Costs indicated for task implementation and/or time for achievement ofrecovery are only estimates and are subject to change. Recovery plans do not necessarily represent the views nor the official positions or approval ofany individuals or agencies involved in the plan formulation, otherthan the U.S. Fish and Wildlife Service. They represent the official position ofthe U.S. -

Bioreactor Cultivation of Zeltnera Beyrichii (Torr
ORIGINAL ARTICLE Rec. Nat. Prod . 7:4 (2013) 266-280 Bioreactor Cultivation of Zeltnera beyrichii (Torr. & A. Gray) Mans.: A Novel Source of Biologically Active Compounds Miloš Radovi ć, Branislav Šiler ∗∗∗, Jasmina Nestorovi ć Živkovi ć, Tijana Banjanac, Suzana Živkovi ć, Miloš Nikoli ć, Marina Sokovi ć and Danijela Miši ć Institute for Biological Research, “Siniša Stanković”, University of Belgrade, Bul. despota Stefana 142, 11060 Belgrade, Serbia (Received February 27, 2013; Revised June 14, 2013; Accepted July 8, 2013) Abstract: With regard to world’s increasing demand for biologically active compounds, a novel source of xanthones and secoiridoid glycosides has been studied. Zeltnera beyrichii (Torr. & A. Gray) Mans., an insufficiently acknowledged North American medicinal plant species, may be considered a pharmacological substitute for commercial C. erythraea Rafn, since it accumulates in aerial parts nearly the same amount of secoiridoid glycosides: swertiamarin, gentiopicrin, and sweroside (13.76, 7.56, and 0.17 mmol per 100 g dry weight, respectively) in plants grown under greenhouse condition, and a considerable amount of xanthones: decussatin and eustomin. Additionally, Z. beyrichii produced as much biomass during cultivation in RITA® temporary immersion bioreactors as greenhouse-grown plants, in a third of the time. Plants grown in bioreactors contained moderate levels of total phenolics and total flavonoids, and possessed modest antioxidant activity and antimicrobial potential against eight bacterial and eight fungal species. Therefore, this species may be highly recommended for cultivation either in natural environment, or in bioreactors under in vitro conditions, for producing compounds of interest of modern pharmacology and food industry. Keywords: Zelnera beyrichii; temporary immersion bioreactors; secoiridoid glycosides; xanthones; antioxidant activity; antimicrobial activity. -
Dwarf Thistle, Cirsi
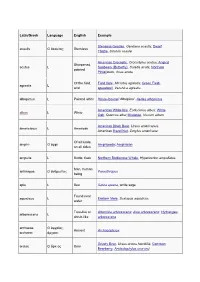
Latin/Greek Language English Example Stemless Gentian, Gentiana acaulis; Dwarf acaulis G ἄκαυλος Stemless Thistle, Cirsium acaule American Crocodile, Crocodylus acutus; Angled Sharpened, acutus L Sunbeam (Butterfly), Curetis acuta; Northern pointed Pintailduck, Anas acuta Of the field, Field Vole, Microtus agrestis; Green Field- agrestis L wild speedwell, Veronica agrestis albopictus L Painted white Hosta fortunei 'Albopicta', Aedes albopictus American White Ibis, Eudocimus albus; White albus L White Oak, Quercus alba; Mistletoe, Viscum album American Black Bear, Ursus americanus; americanus L American American Hazel Nut, Corylus americana Of all kinds, amphi- G ἀμφί Amphipoda; Amphibian on all sides ampulla L Bottle, flask Northern Bottlenose Whale, Hyperoodon ampullatus Man, human anthropos G ἄνθρωπος Paranthropus being apis L Bee Salvia apiana, white sage Found near aquaticus L Eastern Mole, Scalopus aquaticus water Tree-like or Artemisia arborescens; Aloe arborescens; Hydrangea arborescens L shrub-like arborescens archaeos, G ἀρχαῖος, Ancient Archaeopteryx archaeo- ἀρχαιο- Grizzly Bear, Ursus arctos horribilis; Common arctos G ἄρκτος Bear Bearberry, Arctostaphylos uva-ursi argentatus L Silvery Herring Gull, Larus argentatus arthron G ἄρθρον Joint Arthropoda arvensis L In the field Skylark, Alauda arvensis astron, astro-, G ἄστρον, Star Starfish (class), Asteroidea astero- ἀστρο-, ἀστερο- Acer palmatum 'Atropurpureum'; Berberis atropurpureum L Deep purple thunbergii f. atropurpurea Daphne odora 'Aureomarginata'; Taxus aureomarginata -

Health Effects Support Document for Perfluorooctanoic Acid (PFOA)
United States Office of Water EPA 822-R-16-003 Environmental Protection Mail Code 4304T May 2016 Agency Health Effects Support Document for Perfluorooctanoic Acid (PFOA) Perfluorooctanoic Acid – May 2016 i Health Effects Support Document for Perfluorooctanoic Acid (PFOA) U.S. Environmental Protection Agency Office of Water (4304T) Health and Ecological Criteria Division Washington, DC 20460 EPA Document Number: 822-R-16-003 May 2016 Perfluorooctanoic Acid – May 2016 ii BACKGROUND The Safe Drinking Water Act (SDWA), as amended in 1996, requires the Administrator of the U.S. Environmental Protection Agency (EPA) to periodically publish a list of unregulated chemical contaminants known or anticipated to occur in public water systems and that may require regulation under SDWA. The SDWA also requires the Agency to make regulatory determinations on at least five contaminants on the Contaminant Candidate List (CCL) every 5 years. For each contaminant on the CCL, before EPA makes a regulatory determination, the Agency needs to obtain sufficient data to conduct analyses on the extent to which the contaminant occurs and the risk it poses to populations via drinking water. Ultimately, this information will assist the Agency in determining the most appropriate course of action in relation to the contaminant (e.g., developing a regulation to control it in drinking water, developing guidance, or deciding not to regulate it). The PFOA health assessment was initiated by the Office of Water, Office of Science and Technology in 2009. The draft Health Effects Support Document for Perfluoroctanoic Acid (PFOA) was completed in 2013 and released for public comment in February 2014. -

North American Rock Garden Society |
Bulletin of the American Rock Garden Society Volume 50 Number 4 Fall 1992 Cover: Gentiana paradoxa by Rob Proctor of Denver, Colorado Bulletin of the American Rock Garden Society Volume 50 Number 4 Fall 1992 Features Sorting out the Gentians, by Geoffrey Charlesworth 243 Fritillaries of Central Asia, by Josef Slegl 253 Trillium Rescue, by Don L. Jacobs 261 The Story of Fritillaria 'Martha Roderick', by W.H. de Goede 264 New Home for Rock Plants, by Elisabeth Sheldon 265 Eriogonums: Secret of the Dry Garden, by Irma Gourley 271 Preserving Rock Garden Specimens, by Karen Matthews 275 Spontaneity on the Rocks, by Panayoti Kelaidis 285 The Arctic Harebell, by J.S. DeSanto 291 Hunting for Red Helleborus niger, by Will McLewin 295 Departments Plant Portrait: Gentiana paradoxa 276 Awards 299 Books 305 Gentiana algida 242 Bulletin of the American Rock Garden Society Vol. 50(4) Sorting out the Gentians by Geoffrey Charlesworth 1 here are some genera in which tors. It is one of the hallmarks of a many of the species are considered good grower if a large patch can be good alpine plants. Androsace is such produced and maintained year after a genus, and we tend to dismiss the year, but the despair of most of us, who species that are not up to the highest have only occasionally seen a few small standard as not worth growing—for plants in our own gardens and then not instance, A. loctiflora or A. albana. It always with the astonishing color we is a mistake to make such odious associate with the species. -

Phylogeny and Phylogenetic Taxonomy of Dipsacales, with Special Reference to Sinadoxa and Tetradoxa (Adoxaceae)
PHYLOGENY AND PHYLOGENETIC TAXONOMY OF DIPSACALES, WITH SPECIAL REFERENCE TO SINADOXA AND TETRADOXA (ADOXACEAE) MICHAEL J. DONOGHUE,1 TORSTEN ERIKSSON,2 PATRICK A. REEVES,3 AND RICHARD G. OLMSTEAD 3 Abstract. To further clarify phylogenetic relationships within Dipsacales,we analyzed new and previously pub- lished rbcL sequences, alone and in combination with morphological data. We also examined relationships within Adoxaceae using rbcL and nuclear ribosomal internal transcribed spacer (ITS) sequences. We conclude from these analyses that Dipsacales comprise two major lineages:Adoxaceae and Caprifoliaceae (sensu Judd et al.,1994), which both contain elements of traditional Caprifoliaceae.Within Adoxaceae, the following relation- ships are strongly supported: (Viburnum (Sambucus (Sinadoxa (Tetradoxa, Adoxa)))). Combined analyses of C ap ri foliaceae yield the fo l l ow i n g : ( C ap ri folieae (Diervilleae (Linnaeeae (Morinaceae (Dipsacaceae (Triplostegia,Valerianaceae)))))). On the basis of these results we provide phylogenetic definitions for the names of several major clades. Within Adoxaceae, Adoxina refers to the clade including Sinadoxa, Tetradoxa, and Adoxa.This lineage is marked by herbaceous habit, reduction in the number of perianth parts,nectaries of mul- ticellular hairs on the perianth,and bifid stamens. The clade including Morinaceae,Valerianaceae, Triplostegia, and Dipsacaceae is here named Valerina. Probable synapomorphies include herbaceousness,presence of an epi- calyx (lost or modified in Valerianaceae), reduced endosperm,and distinctive chemistry, including production of monoterpenoids. The clade containing Valerina plus Linnaeeae we name Linnina. This lineage is distinguished by reduction to four (or fewer) stamens, by abortion of two of the three carpels,and possibly by supernumerary inflorescences bracts. Keywords: Adoxaceae, Caprifoliaceae, Dipsacales, ITS, morphological characters, phylogeny, phylogenetic taxonomy, phylogenetic nomenclature, rbcL, Sinadoxa, Tetradoxa. -

ISB: Atlas of Florida Vascular Plants
Longleaf Pine Preserve Plant List Acanthaceae Asteraceae Wild Petunia Ruellia caroliniensis White Aster Aster sp. Saltbush Baccharis halimifolia Adoxaceae Begger-ticks Bidens mitis Walter's Viburnum Viburnum obovatum Deer Tongue Carphephorus paniculatus Pineland Daisy Chaptalia tomentosa Alismataceae Goldenaster Chrysopsis gossypina Duck Potato Sagittaria latifolia Cow Thistle Cirsium horridulum Tickseed Coreopsis leavenworthii Altingiaceae Elephant's foot Elephantopus elatus Sweetgum Liquidambar styraciflua Oakleaf Fleabane Erigeron foliosus var. foliosus Fleabane Erigeron sp. Amaryllidaceae Prairie Fleabane Erigeron strigosus Simpson's rain lily Zephyranthes simpsonii Fleabane Erigeron vernus Dog Fennel Eupatorium capillifolium Anacardiaceae Dog Fennel Eupatorium compositifolium Winged Sumac Rhus copallinum Dog Fennel Eupatorium spp. Poison Ivy Toxicodendron radicans Slender Flattop Goldenrod Euthamia caroliniana Flat-topped goldenrod Euthamia minor Annonaceae Cudweed Gamochaeta antillana Flag Pawpaw Asimina obovata Sneezeweed Helenium pinnatifidum Dwarf Pawpaw Asimina pygmea Blazing Star Liatris sp. Pawpaw Asimina reticulata Roserush Lygodesmia aphylla Rugel's pawpaw Deeringothamnus rugelii Hempweed Mikania cordifolia White Topped Aster Oclemena reticulata Apiaceae Goldenaster Pityopsis graminifolia Button Rattlesnake Master Eryngium yuccifolium Rosy Camphorweed Pluchea rosea Dollarweed Hydrocotyle sp. Pluchea Pluchea spp. Mock Bishopweed Ptilimnium capillaceum Rabbit Tobacco Pseudognaphalium obtusifolium Blackroot Pterocaulon virgatum -

Supplementary File 2A Revised
Supplementary file 2A. Differentially expressed genes in aldosteronomas compared to all other samples, ranked according to statistical significance. Missing values were not allowed in aldosteronomas, but to a maximum of five in the other samples. Acc UGCluster Name Symbol log Fold Change P - Value Adj. P-Value B R99527 Hs.8162 Hypothetical protein MGC39372 MGC39372 2,17 6,3E-09 5,1E-05 10,2 AA398335 Hs.10414 Kelch domain containing 8A KLHDC8A 2,26 1,2E-08 5,1E-05 9,56 AA441933 Hs.519075 Leiomodin 1 (smooth muscle) LMOD1 2,33 1,3E-08 5,1E-05 9,54 AA630120 Hs.78781 Vascular endothelial growth factor B VEGFB 1,24 1,1E-07 2,9E-04 7,59 R07846 Data not found 3,71 1,2E-07 2,9E-04 7,49 W92795 Hs.434386 Hypothetical protein LOC201229 LOC201229 1,55 2,0E-07 4,0E-04 7,03 AA454564 Hs.323396 Family with sequence similarity 54, member B FAM54B 1,25 3,0E-07 5,2E-04 6,65 AA775249 Hs.513633 G protein-coupled receptor 56 GPR56 -1,63 4,3E-07 6,4E-04 6,33 AA012822 Hs.713814 Oxysterol bining protein OSBP 1,35 5,3E-07 7,1E-04 6,14 R45592 Hs.655271 Regulating synaptic membrane exocytosis 2 RIMS2 2,51 5,9E-07 7,1E-04 6,04 AA282936 Hs.240 M-phase phosphoprotein 1 MPHOSPH -1,40 8,1E-07 8,9E-04 5,74 N34945 Hs.234898 Acetyl-Coenzyme A carboxylase beta ACACB 0,87 9,7E-07 9,8E-04 5,58 R07322 Hs.464137 Acyl-Coenzyme A oxidase 1, palmitoyl ACOX1 0,82 1,3E-06 1,2E-03 5,35 R77144 Hs.488835 Transmembrane protein 120A TMEM120A 1,55 1,7E-06 1,4E-03 5,07 H68542 Hs.420009 Transcribed locus 1,07 1,7E-06 1,4E-03 5,06 AA410184 Hs.696454 PBX/knotted 1 homeobox 2 PKNOX2 1,78 2,0E-06 -

In Vitro Regeneration and Transformation of Blackstonia Perfoliata
BIOLOGIA PLANTARUM 48 (3): 333-338, 2004 In vitro regeneration and transformation of Blackstonia perfoliata A. BIJELOVIĆ*1, N. ROSIĆ**, J. MILJUŠ-DJUKIĆ***, S. NINKOVIĆ** and D. GRUBIŠIĆ** Institute of Botany, Faculty of Biology, Takovska 43, 11000 Belgrade, Serbia and Montenegro* Institute for Biological Research “Siniša Stanković”, 29. Novembra 142, 11060 Belgrade, Serbia and Montenegro** Institute for Molecular Genetics and Genetic Engineering, Vojvode Stepe 444a, 11000 Belgrade, Serbia and Montenegro*** Abstract In vitro root culture of yellow wort (Blackstonia perfoliata (L.) Huds.) was initiated on Murashige and Skoog (MS) medium. In the presence of benzylaminopurine (BAP) numerous adventitious buds formed, which developed into shoots. Presence of indole-3-butyric acid (IBA) in media significantly decreased number of buds, but increased development of lateral roots. On hormone-free medium shoots successfully rooted and developed flowers and viable seeds that formed another generation. Shoot cultures of B. perfoliata inoculated with suspension of Agrobacterium rhizogenes strain A4M70GUS developed hairy roots at 3 weeks and they were cultured on hormone-free MS medium. Spontaneous shoot regeneration occurred in 3 clones. Additional key words: Agrobacterium rhizogenes, hairy roots, regeneration, root culture. Introduction Blackstonia perfoliata (yellow wort) (L.) Huds. (Chlora 1997a, Menković et al. 1998, Vintehalter and Vinterhalter perfoliata L., Gentiana perfoliata L., Seguiera perfoliata 1998, Mikula and Rybczynski 2001). O. Kuntze), Gentianaceae, is an annual plant, 10 - 60 cm Since B. perfoliata could be used in medicine instead high, with long internodes, triangular leaves, sometimes of Radix Gentianae, this plant can be produced in great narrowing towards the base (Jovanović-Dunjić 1973). It biomass in culture in vitro. -

Centaurium Tenuiflorum (Gentianaceae) New to Arkansas
Singhurst, J.R., J. Keesling, and W.C. Holmes. 2015. Centaurium tenuiflorum (Gentianaceae) new to Arkansas. Phytoneuron 2015-55: 1–2. Published 20 October 2015. ISSN 2153 733X CENTAURIUM TENUIFLORUM (GENTIANACEAE) NEW TO ARKANSAS JASON R. SINGHURST Wildlife Diversity Program Texas Parks and Wildlife Department 4200 Smith School Road Austin, Texas 78744 JIM KEESLING 83 San Juan Way Hot Springs Village, Arkansas 71909 WALTER C. HOLMES Department of Biology Baylor University Waco, Texas 76798-7388 ABSTRACT Centaurium tenuiflorum , a native of Europe, is reported for the first time as naturalized in Arkansas. The species is known to be naturalized in the nearby states of Texas, Oklahoma, Louisiana, and Mississippi as well as in California and Oregon. Based on the following specimens collected in southern Arkansas, we report Centaurium tenuiflorum (Hoffmanns. & Link) Fritsch ex Janch., Figure 1, (Gentianaceae) as new to the spontaneous flora of that state. Arkansas . Miller Co. : E side of Hwy 49, 0.4 mi S of jct with Arkansas Blvd, slope right-of- way, near Texarkana Airport (33° 27' 15.93" N, 94° 0' 34.36" W), 20 May 2015, Singhurst 20995 (BAYLU). Saline Co. : N side of I-30, 0.8 mi W of jct with Hwy 67, right-of-way along access road, W side of the weigh station lane (34° 32' 30.34" N, 92° 38' 37.82" W), 27 May 2015, Keesling 54 (ANHC); W side of Hill Farm Road 0.8 mi from jct with South Reynolds Road and across from Central Arkansas Flying Club Airport (34° 35' 15.12" N, 92° 29' 10.52" W), 6 Jul 2015, Keesling 70 (ANHC).