Fractional Distillation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Extraction of Wax from Sorghum Bran •
i EXTRACTION OF WAX FROM SORGHUM BRAN • 1 HSIEN-WEN HSU <& . ***£/ B. S., National Taiwan University, China, 1951 A THESIS » submitted in partial fulfillment of the requirements for the degree MASTER OF SCIENCE - Department of Chemical Engineering KANSAS STATE COLLEGE OF AGRICULTURE AND APPLIED SCIENCE - 1955 » k Lo i~i ii Q^cu^v^Js TABLE OF CONTENTS INTRODUCTION 1 PREVIOUS WORK 3 MATERIALS 6 GLASS SOXHLET EXTRACTION AND ANALYSIS OF WAX AND OIL IN MISCELLA 7 Procedure of Using Soxhlet Extractor 7 Separation of Wax from Oil by the Acetone Method.. 8 Summary of Results 9 PILOT PLANT EXTRACTION 15 SEPARATION OF WAX AND OIL 16 Effect of Temperature in the Acetone Method 16 Urea Complex Method of Separation. 17 Theory 17 Experimental Procedure 22 Summary of Results 26 COST ESTIMATION OF A WAX RECOVERY PLANT 29 DISCUSSION 30 CONCLUSIONS 35 ACKNOWLEDGMENTS 36 BIBLIOGRAPHY 37 APPENDIX 39 INTRODUCTION i One of the functions of the Agricultural Experiment Station is the never ending search for means of better and fuller utilization of the agri- cultural products that are of particular interest in the state of Kansas. In line with this broad objective, the extraction of economically valuable lipid materials from the sorghum bran has been studied in several theses in the Departments of Chemical Engineering and Chemistry in recent years. Generally the emphasis has b. en placed upon either the chemical and physical properties of these lipides or the equipment design and performance for the solvent ex- traction operation. A brief review of the previous work is presented in the ensuing section. -

The Separation of Three Azeotropes by Extractive Distillation by An-I Yeh A
The separation of three azeotropes by extractive distillation by An-I Yeh A thesis submitted in partial fulfillment of the requirement for the degree of Master of Science in Chemical Engineering Montana State University © Copyright by An-I Yeh (1983) Abstract: Several different kinds of extractive distillation agents were investigated to affect the separation of three binary liquid mixtures, isopropyl ether - acetone, methyl acetate - methanol, and isopropyl ether - methyl ethyl ketone. Because of the small size of the extractive distillation column, relative volatilities were assumed constant and the Fenske equation was used to calculate the relative volatilities and the number of minimum theoretical plates. Dimethyl sulfoxide was found to be a good extractive distillation agent. Extractive distillation when employing a proper agent not only negated the azeotropes of the above mixtures, but also improved the efficiency of separation. This process could reverse the relative volatility of isopropyl ether and acetone. This reversion was also found in the system of methyl acetate and methanol when nitrobenzene was the agent. However, normal distillation curves were obtained for the system of isopropyl ether and methyl ethyl ketone undergoing extractive distillation. In the system of methyl acetate and methanol, the relative volatility decreased as the agents' carbon number increased when glycols were used as the agents. In addition, the oxygen number and the locations of hydroxyl groups in the glycols used were believed to affect the values of relative volatility. An appreciable amount of agent must be maintained in the column to affect separation. When dimethyl sulfoxide was an agent for the three systems studied, the relative volatility increased as the addition rate increased. -

Distillation 65 Chem 355 Jasperse DISTILLATION
Distillation 65 Chem 355 Jasperse DISTILLATION Background Distillation is a widely used technique for purifying liquids. The basic distillation process involves heating a liquid such that liquid molecules vaporize. The vapors produced are subsequently passed through a water-cooled condenser. Upon cooling, the vapor returns to it’s liquid phase. The liquid can then be collected. The ability to separate mixtures of liquids depends on differences in volatility (the ability to vaporize). For separation to occur, the vapor that is condensed and collected must be more pure than the original liquid mix. Distillation can be used to remove a volatile solvent from a nonvolatile product; to separate a volatile product from nonvolatile impurities; or to separate two or more volatile products that have sufficiently different boiling points. Vaporization and Boiling When a liquid is placed in a closed container, some of the molecules evaporate into any unoccupied space in the container. Evaporation, which occurs at temperatures below the boiling point of a compound, involves the transition from liquid to vapor of only those molecules at the liquid surface. Evaporation continues until an equilibrium is reached between molecules entering and leaving the liquid and vapor states. The pressure exerted by these gaseous molecules on the walls of the container is the equilibrium vapor pressure. The magnitude of this vapor pressure depends on the physical characteristics of the compound and increases as temperature increases. In an open container, equilibrium is never established, the vapor can simply leave, and the liquid eventually disappears. But whether in an open or closed situation, evaporation occurs only from the surface of the liquid. -
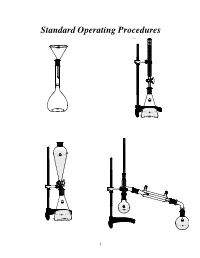
Standard Operating Procedures
Standard Operating Procedures 1 Standard Operating Procedures OVERVIEW In the following laboratory exercises you will be introduced to some of the glassware and tech- niques used by chemists to isolate components from natural or synthetic mixtures and to purify the individual compounds and characterize them by determining some of their physical proper- ties. While working collaboratively with your group members you will become acquainted with: a) Volumetric glassware b) Liquid-liquid extraction apparatus c) Distillation apparatus OBJECTIVES After finishing these sessions and reporting your results to your mentor, you should be able to: • Prepare solutions of exact concentrations • Separate liquid-liquid mixtures • Purify compounds by recrystallization • Separate mixtures by simple and fractional distillation 2 EXPERIMENT 1 Glassware Calibration, Primary and Secondary Standards, and Manual Titrations PART 1. Volumetric Glassware Calibration Volumetric glassware is used to either contain or deliver liquids at a specified temperature. Glassware manufacturers indicate this by inscribing on the volumetric ware the initials TC (to contain) or TD (to deliver) along with the calibration temperature, which is usually 20°C1. Volumetric glassware must be scrupulously clean before use. The presence of streaks or droplets is an indication of the presence of a grease film. To eliminate grease from glassware, scrub with detergent solution, rinse with tap water, and finally rinse with a small portion of distilled water. Volumetric flasks (TC) A volumetric flask has a large round bottom with only one graduation mark positioned on the long narrow neck. Graduation Mark Stopper The position of the mark facilitates the accurate and precise reading of the meniscus. If the flask is used to prepare a solution starting with a solid compound, add small amounts of sol- vent until the entire solid dissolves. -

Alembic Pot Still
ALEMBIC POT STILL INSTRUCTION MANUAL CAN BE USED WITH THE GRAINFATHER OR T500 BOILER SAFETY Warning: This system produces a highly flammable liquid. PRECAUTION: • Always use the Alembic Pot Still System in a room with adequate ventilation. • Never leave the Alembic Pot Still system unattended when operating. • Keep the Alembic Pot Still system away from all sources of ignition, including smoking, sparks, heat, and open flames. • Ensure all other equipment near to the Alembic Pot Still system or the alcohol is earthed. • A fire extinguishing media suitable for alcohol should be kept nearby. This can be water fog, fine water spray, foam, dry powder, carbon dioxide, sand or dolomite. • Do not boil dry. In the event the still is boiled dry, reset the cutout button under the base of the still. In the very unlikely event this cutout fails, a fusible link gives an added protection. IN CASE OF SPILLAGE: • Shut off all possible sources of ignition. • Clean up spills immediately using cloth, paper towels or other absorbent materials such as soil, sand or other inert material. • Collect, seal and dispose accordingly • Mop area with excess water. CONTENTS Important points before getting started ............................................................................... 3 Preparing the Alembic Pot Still ................................................................................................. 5 Distilling a Whiskey, Rum or Brandy .......................................................................................7 Distilling neutral -
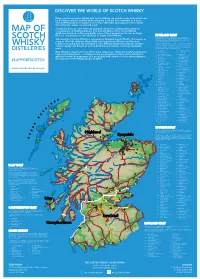
2019 Scotch Whisky
©2019 scotch whisky association DISCOVER THE WORLD OF SCOTCH WHISKY Many countries produce whisky, but Scotch Whisky can only be made in Scotland and by definition must be distilled and matured in Scotland for a minimum of 3 years. Scotch Whisky has been made for more than 500 years and uses just a few natural raw materials - water, cereals and yeast. Scotland is home to over 130 malt and grain distilleries, making it the greatest MAP OF concentration of whisky producers in the world. Many of the Scotch Whisky distilleries featured on this map bottle some of their production for sale as Single Malt (i.e. the product of one distillery) or Single Grain Whisky. HIGHLAND MALT The Highland region is geographically the largest Scotch Whisky SCOTCH producing region. The rugged landscape, changeable climate and, in The majority of Scotch Whisky is consumed as Blended Scotch Whisky. This means as some cases, coastal locations are reflected in the character of its many as 60 of the different Single Malt and Single Grain Whiskies are blended whiskies, which embrace wide variations. As a group, Highland whiskies are rounded, robust and dry in character together, ensuring that the individual Scotch Whiskies harmonise with one another with a hint of smokiness/peatiness. Those near the sea carry a salty WHISKY and the quality and flavour of each individual blend remains consistent down the tang; in the far north the whiskies are notably heathery and slightly spicy in character; while in the more sheltered east and middle of the DISTILLERIES years. region, the whiskies have a more fruity character. -

United States Patent (10) Patent No.: US 9,586,922 B2 Wood Et Al
USOO9586922B2 (12) United States Patent (10) Patent No.: US 9,586,922 B2 Wood et al. (45) Date of Patent: Mar. 7, 2017 (54) METHODS FOR PURIFYING 9,388,150 B2 * 7/2016 Kim ..................... CO7D 307/48 5-(HALOMETHYL)FURFURAL 9,388,151 B2 7/2016 Browning et al. 2007/O161795 A1 7/2007 Cvak et al. 2009, 0234142 A1 9, 2009 Mascal (71) Applicant: MICROMIDAS, INC., West 2010, 0083565 A1 4, 2010 Gruter Sacramento, CA (US) 2010/0210745 A1 8/2010 McDaniel et al. 2011 O144359 A1 6, 2011 Heide et al. (72) Inventors: Alex B. Wood, Sacramento, CA (US); 2014/01 00378 A1 4/2014 Masuno et al. Shawn M. Browning, Sacramento, CA 2014/O1878O2 A1 7/2014 Mikochik et al. (US);US): Makoto N. Masuno,M. s Elk Grove, s 2015/02668432015,0203462 A1 9/20157/2015 CahanaBrowning et etal. al. CA (US); Ryan L. Smith, Sacramento, 2016/0002190 A1 1/2016 Browning et al. CA (US); John Bissell, II, Sacramento, 2016,01681 07 A1 6/2016 Masuno et al. CA (US) 2016/02O7897 A1 7, 2016 Wood et al. (73) Assignee: MICROMIDAS, INC., West FOREIGN PATENT DOCUMENTS Sacramento, CA (US) CN 101475544. A T 2009 (*) Notice: Subject to any disclaimer, the term of this N 1939: A 658 patent is extended or adjusted under 35 DE 635,783 C 9, 1936 U.S.C. 154(b) by 0 days. EP 291494 A2 11, 1988 EP 1049657 B1 3, 2003 (21) Appl. No.: 14/852,306 GB 1220851 A 1, 1971 GB 1448489 A 9, 1976 RU 2429234 C2 9, 2011 (22) Filed: Sep. -

Laboratory Rectifying Stills of Glass 2
» LABORATORY RECTIFYING STILLS OF GLASS 2 By Johannes H. Bruun 3 and Sylvester T. Schicktanz 3 ABSTRACT A complete description is given of a set of all-glass rectifying stills, suitable for distillation at pressures ranging from atmospheric down to about 50 mm. The stills are provided with efficient bubbling-cap columns containing 30 to 60 plates. Adiabatic conditions around the column are maintained by surrounding it with a jacket provided with a series of independent electrical heating units. Suitable means are provided for adjusting or maintaining the reflux ratio at the top of the column. For the purpose of conveniently obtaining an accurate value of the true boiling points of the distillates a continuous boiling-point apparatus is incor- porated in the receiving system. An efficient still of the packed-column type for distillations under pressures less than 50 mm is also described. Methods of operation and efficiency tests are given for the stills. CONTENTS Page I. Introduction 852 II. General features of the still assembly 852 III. Still pot 853 IV. Filling tube and heater for the still pot 853 V. Rectifying column 856 1. General features 856 2. Large-size column 856 3. Medium-size column 858 4. Small-size column 858 VI. Column jacket 861 VII. Reflux regulator 861 1. With variable reflux ratio 861 2. With constant reflux ratio 863 VIII. The continuous boiling-point apparatus 863 IX. Condensers and receiver 867 X. The mounting of the still 867 XI. Manufacture and transportation of the bubbling-cap still 870 XII. Operation of the bubbling-cap still 870 XIII. -

Hydro-Distillation and Steam Distillation from Aromatic Plants
Hydro-distillation and steam distillation from aromatic plants Sudeep Tandon Scientist Chemical Engineering Division CIMAP, Lucknow HISTORY Written records of herbal distillation are found as early as the first century A.D., and around 1000 A.D., the noted Arab physician and naturalist Ibn Sina also known as Avicenna described the distillation of rose oil from rose petals The ancient Arabian people began to study the chemical properties of essential oils & developed and refined the distillation process Europeans began producing essential oils in the 12th century 1 DISTILLATION ? A process in which a liquid or vapour mixture of two or more substances is separated into its component fractions of desired purity, by the application and removal of heat. In simple terms distillation of aromatic herbs implies vaporizing or liberating the oils from the trichomes / plant cell membranes of the herb in presence of high temperature and moisture and then cooling the vapour mixture to separate out the oil from water. It is the most popular widely used and cost effective method in use today for producing majority of the essential oils throughout the world Distillation is an art and not just a “Chemical" process that is reliant upon many factors for successful quality oil production. BASIC SCIENTIFIC PRINCIPLES INVOLVED IN THE PROCESS To convert any liquid into a vapour we have to apply energy in form of heat called as latent heat of vaporization A liquid always boils at the temperature at which its vapour pressure equals the atmospheric / surrounding pressure For two immiscible liquids the total vapour pressure of the mixture is always equal to the sum of their partial pressures The composition of the mixture will be determined by the concentration of the individual components into its partial pressure As known the boiling point of most essential oil components exceeds that of water and generally lies between 150 – 300oC 2 If a sample of an essential oil having a component ‘A’ having boiling point for example 190oC and the boiling point of the water is 100oC. -

Building a Home Distillation Apparatus
BUILDING A HOME DISTILLATION APPARATUS A Step by Step Guide Building a Home Distillation Apparatus i BUILDING A HOME DISTILLATION APPARATUS Foreword The pages that follow contain a step-by-step guide to building a relatively sophisticated distillation apparatus from commonly available materials, using simple tools, and at a cost of under $100 USD. The information contained on this site is directed at anyone who may want to know more about the subject: students, hobbyists, tinkers, pure water enthusiasts, survivors, the curious, and perhaps even amateur wine and beer makers. Designing and building this apparatus is the only subject of this manual. You will find that it confines itself solely to those areas. It does not enter into the domains of fermentation, recipes for making mash, beer, wine or any other spirits. These areas are covered in detail in other readily available books and numerous web sites. The site contains two separate design plans for the stills. And while both can be used for a number of distillation tasks, it should be recognized that their designs have been optimized for the task of separating ethyl alcohol from a water-based mixture. Having said that, remember that the real purpose of this site is to educate and inform those of you who are interested in this subject. It is not to be construed in any fashion as an encouragement to break the law. If you believe the law is incorrect, please take the time to contact your representatives in government, cast your vote at the polls, write newsletters to the media, and in general, try to make the changes in a legal and democratic manner. -

Distillation of Essential Oils1
WEC310 Distillation of Essential Oils1 Elise V. Pearlstine 2 A short history of essential oils many industries and in new applications as awareness of the benefit of naturally derived products grows. Essential oils are volatile, aromatic oils obtained from plants and used for fragrance, flavoring, and health and beauty applications. Historically, aromatic plants provided important ingredients for perfumes, incense, and cosmetics. They have also been used for ritual purposes and in cooking and medicine. Egyptians used aromatic plant materials to preserve mummies, the Ayurvedic literature of India includes many references to scented substances, ancient Chinese herbalists valued them for their curative properties, and royalty used rare aromatics to perfume themselves and their surroundings. Distillation became an important An eighteenth century still from an old method of obtaining the healing and fragrant Figure 1. monograph by Gildemeister. components of various plants and was well-studied beginning in the 18th and continuing in the 19th Plant anatomy and structure as they centuries (Figure 1). In the 1900s, during the time of relate to essential oil production the industrial revolution, component parts of many essential oils were identified. These components An essential oil is the volatile material derived could then be synthesized for use in perfume and from plant material by a physical process. The plant flavor industries. The art of using essential oils material is usually aromatic and of a single botanical declined during this time but experienced a re-birth in species and form; some essential oil plants have a Europe with aromatherapy later in the century. In different chemical makeup depending on the variety recent years, the use of essential oils has increased in of plant, and the essential oils are correspondingly unique. -

A Guide to Stills & Distilling
A GUIDE TO STILLS & DISTILLING LEADERS IN HOME DISTILLING AND FILTRATION SYSTEMS THAT MEET AND EXCEED EVEN COMMERCIAL STANDARDS. www.stillspirits.com Reorder: 72648 V1 Reorder: With over 25 years in the Air Still A compact, sleek stainless steel still for those who want a no fuss way market, Still Spirits is a leader of producing 1 L (1 US qt) of 40% ABV spirit at a time. Distils alcohol at 60% strength. Used with a small 10 L (2.6 US Gal) fermenter making this in home distilling and filtration system easy to move and operate from any kitchen, boat or campervan. systems that meet and exceed AVERAGE USE even commercial standards. DIFFICULTY Basic CAPACITY 4 L (1 US Gal) The range features: TIME 7- 10 day Fermentation Premium spirits with equivalent commercial quality Approx 2 hours distilling per 1 L (1 US qt) of 40% alcohol at a fraction of the price. YIELD 2 L (2 US qt) of 40% ABV alcohol per 8 L Homemade spirits in as little as 7 days. (2 US Gal) wash Distilled alcohol from a wash made simply using sugar, PURITY Distils at 60% ABV (120 US proof) before being watered down to 40% (80 US proof) yeast, carbon and drinkable water. Homemade spirit flavoured with your choice of Still Spirits spirit or liqueur essences. QUICK GUIDE TO DISTILLING FEATURES WITH THE AIR STILL 1. Wash – Make an 8 L (2 US Gal) wash by • Easy to operate, set up and use. mixing water, sugar, carbon and yeast With no hoses or complicated in the fermenter.