PROGRESS of CHEMISTRY I Ł '
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

A Wet Process for the Extraction of Mercury
Scholars' Mine Professional Degree Theses Student Theses and Dissertations 1916 A wet process for the extraction of mercury Robert Glenn Sickly Follow this and additional works at: https://scholarsmine.mst.edu/professional_theses Part of the Metallurgy Commons Department: Recommended Citation Sickly, Robert Glenn, "A wet process for the extraction of mercury" (1916). Professional Degree Theses. 140. https://scholarsmine.mst.edu/professional_theses/140 This Thesis - Open Access is brought to you for free and open access by Scholars' Mine. It has been accepted for inclusion in Professional Degree Theses by an authorized administrator of Scholars' Mine. This work is protected by U. S. Copyright Law. Unauthorized use including reproduction for redistribution requires the permission of the copyright holder. For more information, please contact [email protected]. "/389 A '.vET PROCESS FOR THE EXTRACTION OF MERCURY. ROBERT GLENN SICKLY. '~~****** A THESIS subl!li tted to the facul t Jr of the SCHOOL OF lnIN}~S AND METALI.. URGY O~' THE U:nlv~RSITY OF I~ISSOURI in partial fulfillment of the work required for the Degree of METALJJURGICAL :S:NGINEER. Cobalt, Ont. Canada. 1916. ********* Approved by Professor 0 r~· w-e-r-~r~~J#/d' £7' 11/6 TABLE OF COIrTE1:ITS. Subject. Pa.ge. Introduction ------------------------~---------- 2. The nature o~ the mercury losses --------------- 2. The chemical loss o~ mercury ------------------- 3. The mercury process ---------------------------- 5. Flow-sheet ------------------------------------- 7. Chemistry of the process ----------------------- 7. hlethod of dissolving the mercury --------------- 8: Various methods of precipitation --------------- 16. Aluminium precipitation ------------------------ 19. Curve on aluminium precipitation ----------- opp. 21. Experimental plant installation ---------------- 23. Curves on operation of experimental plant --GpP. -

THE DETERMINATION of MERCURY SPECIES in ENVIRONMENTAL and BIOLOGICAL SAMPLES (Technical Report)
Pure & Appl. Chem., Vol. 70, No. 8, pp. 1585-1615, 1998. Printed in Great Britain. 0 1998 IUPAC INTERNATIONAL UNION OF PURE AND APPLIED CHEMISTRY ANALYTICAL CHEMISTRY DIVISION COMMISSION OF MICROCHEMICAL TECHNIQUES AND TRACE ANALYSIS WORKING GROUP ON SPECIATION THE DETERMINATION OF MERCURY SPECIES IN ENVIRONMENTAL AND BIOLOGICAL SAMPLES (Technical Report) Prepared for publication by MASATOSHI MORITA", JUN YOSHINAGA" AND JOHN S. EDMONDST *NationalInstitute for Environmental Studies, 16-2 Onogawa, Tsukuba, Ibaraki 305, Japan; Western Australian Marine Research Laboratories, PO Box 20, North Beach, WA 6020, Australia Republication or reproduction of this report or its storage and/or dissemination by electronic means is permitted without the need for formal IUPAC permission on condition that an acknowledgement, with full reference to the source along with use of the copyright symbol 0,the name IUPAC and the year of publication are prominently visible. Publication of a translation into another language is subject to the additional condition of prior approval from the relevant IUPAC National Adhering Organization. 1585 1586 COMMISSION ON MICROCHEMICAL TECHNIQUES AND TRACE ANALYSIS The determination of mercury species in environmental and biological samples (Technical Report) Abstract: Mercury is released into the hydrosphere, atmosphere and biosphere as a consequence of natural and anthropogenic processes. It is cycled in the environment and undergoes transformations of its chemical forms. Although the number of chemical species encountered in environmental and biological samples is not large, the range of matrices and the toxicological significance of mercury, with a consequent need for ultra-trace determination, have resulted in a considerable analytical literature. Sensitive and selective methods for mercury determination, micro methods for the determination of mercury species, the occurrence of mercury species in the various environmental compartments, and analytical considerations, including the availability of certified reference materials, are reviewed. -

Introduction Cover
Introduction Cover TM Best Practices for Creating High Performance Healing Environments Version 2.2 Operations Section, 2008 Revision December 2008 Title Page Convener: Founding Sponsors: Other Sponsors: Table of Contents Overview 1 – Introduction 2 – Checklist – Operations 3 – Credit Summary – Operations Operations Credits 4 – Integrated Operations & Education 5 – Sustainable Sites Management 6 – Transportation Operations 7 – Facilities Management 8 – Chemical Management 9 – Waste Management 10 – Environmental Services 11 – Food Service 12 – Environmentally Preferable Purchasing 13 – Innovation in Operations Release for Public Use The Green Guide for Health Care (Green Guide), a project of the Center for Maximum Potential Building Systems and Practice Greenhealth, is released for public use in PDF format. All replication in whole or in part must reference the Green Guide and include the limitations on its use described herein. The Green Guide is an open source document that is provided at no charge. Material contained within the Green Guide may not be used by or as part of a for-profit enterprise (for sale or as a component of an educational program) in which attendees are charged fees without the express permission of the Green Guide for Health Care Co- Coordinators. Portions of Green Guide for Health Care-Operations Version 2.2 are borrowed, with permission, from the U.S. Green Building Council’s LEED for Existing Buildings: Operations and Maintenance, January 2008 and LEED for Healthcare public comment draft, November 2007. This 2008 second public comment draft release of Green Guide for Health Care-Operations Version 2.2 represents a major revision of the Green Guide v2.2 Operations section. -

United States Patent Office
Patented Mar. 15, 1949 2.464,203 UNITED STATES PATENT OFFICE 2,464,203 MANUEFACTURE OF DENOESTROL Walace Fraak Short and Gordon van Hobday, Nottingham, England, assignors to Boots Pure Frug Company inited, Nottinghain, England, a coapaay of Great Britain No Drawing. Application May 10, 1944, Serial No. 534,988. In Great Britain May 21, 1943 18 Claims. (C. 260-470) 2 This invention relates to improved processes for an amalgan in an aqueous alkaline solution. the manufacture of y8-bis-(4-hydroxyphenyl)- Preferred anaigans are sodium amalgam and hexane-y&-diol and of the dihydroxy compound potassium amalgam but other amalgams may be ?y8-bis-4-hydroxyphenyl-A56-hexadiene obtained used. from it by dehydration. 5 When the reduction is effected with the use of The latter (known as “Dienoestrol, B. M. J. an analgarin in an alkaline Solution the isolation 1942, 2, page 256) is of importance because it of the pinacol formed may be effected by known possesses oestrogenic propertie5. methods of separation but it is preferred to use It has previously been proposed to reduce the acylation in ethod previously described. p-hydroxypropiophenone to the pinacol, y?-bis O A further object of the invention is to provide (4-hydroxyphenyl)-hexane-yô-diol with the use an improved process for the production of dienoes of aluminium amalgam in moist ether (Dodds, trol from the aforesaid pinacol. Golberg, Lawson and Robinson. Proc. Roy. Hitherto, the pinaco) has been converted into Soc., 1939, B. vol. 127, 140-167) and an electro dienoestrol by treatment with a mixture of acetyl lytic reduction process has also been described in 15 chloride and acetic anhydride (Dodds et al. -

Chemistry 1 Stokiometry Contents
chemistry 1 stokiometry Contents 1 John Dalton 1 1.1 Early life ................................................ 1 1.2 Early careers .............................................. 1 1.3 Scientific contributions ........................................ 1 1.3.1 Meteorology ......................................... 1 1.3.2 Colour blindness ....................................... 1 1.3.3 Measuring mountains in the Lake District .......................... 2 1.3.4 Gas laws ............................................ 2 1.3.5 Atomic theory ......................................... 2 1.3.6 Atomic weights ........................................ 3 1.3.7 Other investigations ...................................... 3 1.3.8 Experimental approach .................................... 4 1.4 Other publications ........................................... 4 1.5 Public life ............................................... 4 1.6 Personal life .............................................. 5 1.7 Disability and death .......................................... 5 1.8 Legacy ................................................. 5 1.9 See also ................................................ 6 1.10 References ............................................... 6 1.11 Sources ................................................ 7 1.12 External links ............................................. 8 2 Atomic theory 9 2.1 History ................................................. 9 2.1.1 Philosophical atomism .................................... 9 2.1.2 Dalton ............................................ -

Co(Dmgh)2Py
HDE/PhD/2/80 UNIVERSITY OF LONDON GENERAL INSTRUCTIONS FOR APPOINTMENT OF EXAMINERS AND FOR CONDUCT OF EXAMINATIONS FOR PH.D. FOR INTERNAL STUDENTS 1. Appointment of Examiners 1.1 General 1.1.1. At least one of the examiners for each candidate shall, whenever practicable, have had experience in examining for the Ph.D. Degree of the University. 1.1.2 If the examiners appointed do not include the Supervisor (or either of two Joint Supervisors) under whom the research has been carried out, then: (a) The Supervisor (or one of two Joint Supervisors) shall be invited to attend the oral examination of his Ph.D. candidate as an observer without the right to question the candidate or participate in any way (see also 3.5. below) (b) The examiners may, at their discretion, consult the Supervisor before completing their Report, particularly if they have doubts relating to the appropriate recommendation to make. (c) The Supervisor or Joint Supervisor(s) shall be entitled to receive a copy of the examiners' Report on request for his own information only. Any Report so given is issued in the strictest confidence to the Supervisor or Joint Supervisor(s) concerned and may not be passed to any third party. 1.1.3. Examiners are advised that all matters relating to the examination must be treated as confidential. Examiners are not permitted to divulge the content of previously unpublished material contained in a candidate's thesis until such time as any restrictions on access to the thesis, which may have been granted by the Academic Council, have been removed. -

Kinetic and Mass-Spectrometric Studies of Atmospherically Relevant Mercury-Bromine Chemistry Elise-Andrée Guérette Department
Kinetic and mass-spectrometric studies of atmospherically relevant mercury-bromine chemistry Elise-Andrée Guérette Department of Chemistry McGill University Montréal, Québec, Canada October 2010 A thesis submitted to McGill University in partial fulfilment of the require ments of the degree of Master of Science ©Elise-Andrée Guérette, 2010. All Rights Reserved. Acknowledgements My thanks go out to Dr. Daniel Deeds for his help with everything having to do with atmospheric pressure chemical ionization mass spectrometry. Working with you was always a pleasure. Alain Tessier from CBAMS at Concordia Univer sity was immensely helpful and creative in his problem solving. Thanks also to Graydon Snider for helpful discussions and helping me get started with the kinetic work. Edward Hudson, Nermin Eltouny, Daniel Deeds, Graydon Snider and Ma hamud Subir all proofread this thesis and provided helpful comments. I would also like to extend grateful thanks to my supervisor, Parisa Ariya, for her understanding and patience, and for providing me with this opportunity. Thanks also for reading this thesis before its submission and for providing valu able comments. Fred Kluck, Rick Rossi and George Kopp from the Department of Chemistry deserve mention for their help building and repairing things, often at short notice. Thanks also to FQRNT, the Department of Chemistry at McGill, and the Dr. and Mrs. Milton Leong fund for providing personal financial assistance during the course of my studies. Thanks last, but perhaps most of all, to my spouse, Craig Titchener, who stood by me every step of the way. Thank you. II Abstract Bromine radicals are thought to play an important role in the chemistry of atmospheric elemental mercury, however uncertainties remain concerning the rate and the mechanism of the reaction. -

British Chemical Abstracts ------A.-Pure Chemistry
BRITISH CHEMICAL ABSTRACTS ------------------ ------------- A.-PURE CHEMISTRY OCTOBER, 1934. General, Physical, and Inorganic Chemistry. o Origin of anomalous displacements in the 46, 124—129).—Radiations corresponding with energy Stark effect of hydro gen. W. S t e u b i n g and P. differences between terms have been found, and new Ja k el (Z. Physik, 1934, 90, 112—132). lines of Kr ix and Xe ix are predicted. N. M. B. A. B. D. C. Spectrum of nickel hydride. A. G. G a y d o n and Demonstration of the axiality of light emission R. W . B. P e a r s e (Naturę, 1934, 134, 287).—Details of the ultra-violet hydrogen lines 1025 and of a spectrum attributed to the mol. NiH are given. 1215 A. R. F r e r i c h s and H. B o m k e (Physikal. Z., L. S. T. 1934, 35, 549—551).—For the 1025 A. line, with Spectrum of zinc. L. B l o c h and E. B l o c h (J. parallel emission, the ratio of the intensity of the long- Phys. Radium, 1934, [vii], 5, 289—298).—The spark wave to that of the short-wave component is 1 :2 . spectrum in vac. revealcd the new Zn iv order of < An approx. val. of 1:1-5 is obtained for opposite 120 lines in the rangę 4000—2500 A. The high- emission. For the 1215 A. line, the components have freąuency electrodeless discharge allowed the separ- the same intensity in both parallel and opposite ation of the orders Zn n, gm ng < 50 new lines, Zn i i i , emission. -

Index to USP 39–NF 34 the Following Index Is for Convenience and Informational Use Only and Shall Not Be Used for Interpretive
Index to USP 39–NF 34 The following Index is for convenience and informational use only and shall not be used for interpretive purposes. In addition to official articles, this Index may also include items recently omitted from the USP–NF in the indicated Book or Supplement. The requirements stated in the General Notices and Requirements section of the USP–NF apply to all articles recognized in the USP–NF and to all general chapters unless specifically stated otherwise. Although this revision (USP 39–NF 34) is generally official beginning May 1, 2016; particular provisions may indicate another earlier or later official date. In addition, the monographs and general chapters listed in this Index may reference other general chapter specifications. The articles listed in this Index are not intended to be autonomous standards and should only be interpreted in the context of the entire USP–NF publication. For the most current version of the USP–NF please see the USP–NF Online Combined Index to USP 39 and NF 34 Abaca-Acety I-1 Combined Index to USP 39 and NF 34, Volumes 1±4 Page citations refer to the pages of Volumes 1, 2, 3, and 4 of USP 39±NF 34. This index is repeated in its entirety in each volume. 1±2280 Volume 1 2281±4454 Volume 2 4455±6446 Volume 3 6447±7608 Volume 4 Numbers in angle brackets such as 〈421〉 refer to chapter numbers in the General Chapters section. and (salts of) chlorpheniramine, and tramadol hydrochloride tablets, 2323, A dextromethorphan, and 2323 pseudoephedrine, oral powder Acetanilide, 2084 Abacavir containing at -

Protecting Groups Third Edition by Philip J. Kocieński
Protecting Groups Third Edition by Philip J. Kocieński Protecting Groups Third Edition by Philip J. Kocieński Index acetal exchange 126, 136, 154, 160, 293, N-adenylated S-methyl-l-cysteine sulfox- 316, 566 imine 516 acetalisation 59–60, 64 Agelastatin A 508, 555 N,N-acetals 98, 369–371, 388 Agrocin 84 477 N,O-acetals 96–99, 173–179, 232, AI-77-B 576 561–568, 626–630 alanine 552 O,O-acetals 5–7, 11, 50–77, 101, 120–160, Albomycin 595 285–320, 342, 426–430 aldol reaction, directed 26 acetamidomethyl 372, 381, 384 alkene 516 acetate esters 3, 168, 206, 325, 334, 338 alkenes, terminal 55 acetate migration 196, 336 O-alkyl nucleophilic scission 399 80% acetic acid 595 alkylammonium formate 467 acetic acid 5–6, 139–140, 156, 175–176, alkylation 39, 87, 94, 370 190, 196, 200, 208, 216, 265, 269, 315, alkylation, asymmetric 411 318, 341, 343, 461, 529, 564–565, 582 S-alkylation 7 acetic acid in ether 599 alkyllithium reagents 137 acetic acid in refluxing 2-propanol 404 allene 70 acetic anhydride 156, 238, 298, 323, 497, Allopumiliotoxin 339A 245 559 Allosamidin 557 acetonides 202 allyl alcohol 606 a-acetoxy 92 allyl bromide 595 p-acetoxybenzyloxycarbonyl (AcOZ) group allyl carbamate 39 518 allyl chloroformate 284, 346, 423, 528 acetoxymethyl acetal 156 allyl esters 12, 37, 422, 517, 526 acetoxymethyl ether 157 allyl ethers 12–13, 39, 246, 276, 278, 422 2-(acetoxymethyl)benzamides 501 allyl isopropenyl dicarbonate 423 N-acetyl neuraminic acid 4, 177, 258, 330 p-allyl palladium 12, 418, 422, 594–595, 2-acetyldimedone 606 606–607 acetylene 490 -
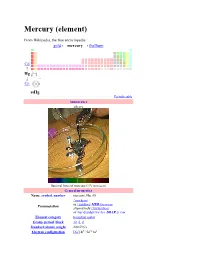
Mercury (Element)
Mercury (element) From Wikipedia, the free encyclopedia gold ← mercury → thallium Cd ↑ Hg ↓ Cn 80Hg Periodic table Appearance silvery Spectral lines of mercury (UV not seen) General properties Name, symbol, number mercury, Hg, 80 /ˈmɜrkjəri/ or /ˈmɜrkəri/ MER-k(y)ə-ree Pronunciation alternatively /ˈkwɪksɪlvər/ or /haɪˈdrɑrdʒɨrəm/ hye-DRAR-ji-rəm Element category transition metal Group, period, block 12, 6, d Standard atomic weight 200.59(2) 14 10 2 Electron configuration [Xe] 4f 5d 6s Electrons per shell 2, 8, 18, 32, 18, 2 (Image) Physical properties Phase liquid Density (near r.t.) 13.534 g·cm−3 Melting point 234.32 K, -38.83 °C, -37.89 °F Boiling point 629.88 K, 356.73 °C, 674.11 °F Critical point 1750 K, 172.00 MPa −1 Heat of fusion 2.29 kJ·mol −1 Heat of vaporization 59.11 kJ·mol −1 −1 Molar heat capacity 27.983 J·mol ·K Vapor pressure P (Pa) 1 10 100 1 k 10 k 100 k at T (K) 315 350 393 449 523 629 Atomic properties 4, 2 (mercuric), 1 (mercurous) Oxidation states (mildly basic oxide) Electronegativity 2.00 (Pauling scale) −1 Ionization energies 1st: 1007.1 kJ·mol 2nd: 1810 kJ·mol−1 3rd: 3300 kJ·mol−1 Atomic radius 151 pm Covalent radius 132±5 pm Van der Waals radius 155 pm Miscellanea Crystal structure rhombohedral [1] Magnetic ordering diamagnetic Electrical resistivity (25 °C) 961nΩ·m −1 −1 Thermal conductivity 8.30 W·m ·K −1 −1 Thermal expansion (25 °C) 60.4 µm·m ·K −1 Speed of sound (liquid, 20 °C) 1451.4 m·s CAS registry number 7439-97-6 Most stable isotopes Main article: Isotopes of mercury iso NA half-life DM DE (MeV) DP 194 -

Download the Full Report Pdf, 1.6 MB, Opens in New Window
VKM Report 2016: 63 Antimicrobial resistance due to the use of biocides and heavy metals: a literature review Opinion of the Panel on Microbial Ecology of the Norwegian Scientific Committee for Food Safety Report from the Norwegian Scientific Committee for Food Safety (VKM) 2016:63 Antimicrobial resistance due to the use of biocides and heavy metals: a literature review Opinion of the Panel Panel on Microbial Ecology of the Norwegian Scientific Committee for Food Safety 09.12.2016 ISBN: 978-82-8259-253-6 Norwegian Scientific Committee for Food Safety (VKM) Po 4404 Nydalen N – 0403 Oslo Norway Phone: +47 21 62 28 00 Email: [email protected] www.vkm.no www.english.vkm.no Suggested citation: VKM. (2016). Antimicrobial resistance due to the use of biocides and heavy metals: a literature review Scientific Opinion on the Panel Panel on Microbial Ecology of the Norwegian Scientific Committee for Food Safety, ISBN: 978-82-8259-253-6, Oslo, Norway. VKM Report 2016: 63 Antimicrobial resistance due to the use of biocides and heavy metals: a literature review Authors preparing the draft opinion Arne Tronsmo (chair), Tor Gjøen, Henning Sørum, and Siamak Yazdankhah (VKM staff) Assessed and approved The opinion has been assessed and approved by the Panel on Microbial Ecology. Members of the panel are: Ida Skaar (chair), Tor Gjøen, Jacques Godfroid, Anders Jelmert, Jörn Klein Arinze Okoli, Arne Tronsmo, og Bjørnar Ytrehus. (Panel members in alphabetical order after chair of the panel) Acknowledgment The Norwegian Scientific Committee for Food Safety (Vitenskapskomiteen for mattrygghet, VKM) has appointed a working group consisting of both VKM members and external experts to answer the request from the Norwegian Food Safety Authority/Norwegian Environment Agency.