Long QT Syndrome Testing
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Non Commercial Use Only
Cardiogenetics 2017; volume 7:6304 Sudden death in a young patient with atrial fibrillation Case Report Correspondence: María Angeles Espinosa Castro, Inherited Cardiovascular Disease A 22-year-old man suffered a sudden Program, Cardiology Department, Gregorio María Tamargo, cardiac arrest without previous symptoms Marañón Hospital, Dr. Esquerdo, 46, 28007, María Ángeles Espinosa, while he was at rest, waiting for a subway Madrid, Spain. Víctor Gómez-Carrillo, Miriam Juárez, train. Cardiopulmonary resuscitation was Tel.: +34.91.586.82.90. immediately started using an Automated E-mail: [email protected] Francisco Fernández-Avilés, External Defibrillation that identified the Raquel Yotti Key words: KCNQ1; mutation; channelopa- presence of ventricular fibrillation and thy; sudden cardiac death; atrial fibrillation. Inherited Cardiovascular Disease delivered a shock. Return of spontaneous Program, Cardiology Department, circulation was achieved after three Contributions: MT, acquisition and interpreta- Gregorio Marañón Hospital, Madrid, attempts, being atrial fibrillation (AF) the tion of data for the work, ensuring that ques- Spain patient’s rhythm at this point (Figure 1). tions related to the accuracy or integrity of any He was admitted to our Cardiovascular part of the work is appropriately investigated Intensive Care Unit and therapeutic and resolved; MAE, conception of the work, hypothermia was performed over a period critical revision of the intellectual content, final approval of the version to be published, Abstract of 24 h. After completing hypothermia, ensuring that questions related to the accuracy rewarming, and another 24 h of controlled of any part of the work is appropriately inves- Sudden cardiac death (SCD) in young normothermia the patient awakened with no tigated and resolved; VG-C, acquisition and patients without structural heart disease is residual neurologic damage. -
Long QT Syndrome Testing
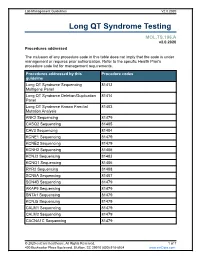
Lab Management Guidelines V2.0.2020 Long QT Syndrome Testing MOL.TS.196.A v2.0.2020 Procedures addressed The inclusion of any procedure code in this table does not imply that the code is under management or requires prior authorization. Refer to the specific Health Plan's procedure code list for management requirements. Procedures addressed by this Procedure codes guideline Long QT Syndrome Sequencing 81413 Multigene Panel Long QT Syndrome Deletion/Duplication 81414 Panel Long QT Syndrome Known Familial 81403 Mutation Analysis ANK2 Sequencing 81479 CASQ2 Sequencing 81405 CAV3 Sequencing 81404 KCNE1 Sequencing 81479 KCNE2 Sequencing 81479 KCNH2 Sequencing 81406 KCNJ2 Sequencing 81403 KCNQ1 Sequencing 81406 RYR2 Sequencing 81408 SCN5A Sequencing 81407 SCN4B Sequencing 81479 AKAP9 Sequencing 81479 SNTA1 Sequencing 81479 KCNJ5 Sequencing 81479 CALM1 Sequencing 81479 CALM2 Sequencing 81479 CACNA1C Sequencing 81479 © 2020 eviCore healthcare. All Rights Reserved. 1 of 7 400 Buckwalter Place Boulevard, Bluffton, SC 29910 (800) 918-8924 www.eviCore.com Lab Management Guidelines V2.0.2020 What is Long QT syndrome Definition Long QT Syndrome (LQTS) is caused by mutations in a number of genes, most of which are related to the functioning of sodium or potassium ion channels in the heart.1 Testing may offer prognostic information in some cases, as specific genes and even specific mutations within those genes may have some correlation to risk for sudden death, effectiveness of beta-blocker therapy, and preventive strategies.1-4 Signs and symptoms of long QT syndrome (LQTS) are variable, but may include a prolonged QT interval on an electrocardiogram, torsades de pointes, syncope, seizures, cardiac arrest, and sudden cardiac death.1,2 Many patients with LQTS can be largely asymptomatic, with cardiac arrest or sudden cardiac death as the first and only symptom. -

Cardiac Arrest: an Important Public Health Issue
Cardiac Arrest: An Important Public Health Issue Cardiac arrest is a public health issue with widespread incidence and severe impact on human health and well-being. There are several recommended strategies for prevention and control. Incidence Impact In 2015, approximately Mortality: 357,000 people experienced 70%–90% out-of-hospital cardiac arrest (OHCA) in the United Approximately 70%–90% of individuals with OHCA die States. before reaching the hospital. Approximately 209,000 Morbidity: Those who survive cardiac arrest are people are treated for in- likely to suffer from injury to the brain and hospital cardiac arrest nervous system and other physical ailments. (IHCA) each year. Additionally, nearly half of OHCA survivors suffer psychological distress such as anxiety, post traumatic stress disorder, and depression. Economic Impact Societal Cost: The estimated burden to society of death from cardiac arrest is 2 million years of life lost for men and 1.3 million years for women, greater than estimates for all individual $ cancers and most leading causes of death. Prevention Early intervention by CPR and defibrillation:Early, high-quality CPR, including compression only CPR, and use of automated external defibrillators (AEDs) immediately following cardiac arrest can reduce morbidity and save lives. Clinical prevention: For Other early interventions: Depending on the patients at high risk, cause of the cardiac arrest, other interventions implantable cardioverter such as cold therapy and administering antidote defibrillators and to toxin-related cardiac arrest can reduce pharmacologic therapies mortality and long-term side effects. can prevent cardiac arrest. What Is Public Health’s Role in Cardiac Arrest? The public health community can implement strategies to prevent and control cardiac arrest. -

Drugs and Life-Threatening Ventricular Arrhythmia Risk: Results from the DARE Study Cohort
Open Access Research BMJ Open: first published as 10.1136/bmjopen-2017-016627 on 16 October 2017. Downloaded from Drugs and life-threatening ventricular arrhythmia risk: results from the DARE study cohort Abigail L Coughtrie,1,2 Elijah R Behr,3,4 Deborah Layton,1,2 Vanessa Marshall,1 A John Camm,3,4,5 Saad A W Shakir1,2 To cite: Coughtrie AL, Behr ER, ABSTRACT Strengths and limitations of this study Layton D, et al. Drugs and Objectives To establish a unique sample of proarrhythmia life-threatening ventricular cases, determine the characteristics of cases and estimate ► The Drug-induced Arrhythmia Risk Evaluation study arrhythmia risk: results from the the contribution of individual drugs to the incidence of DARE study cohort. BMJ Open has allowed the development of a cohort of cases of proarrhythmia within these cases. 2017;7:e016627. doi:10.1136/ proarrhythmia. Setting Suspected proarrhythmia cases were referred bmjopen-2017-016627 ► These cases have provided crucial safety by cardiologists across England between 2003 and 2011. information, as well as underlying clinical and ► Prepublication history for Information on demography, symptoms, prior medical and genetic data. this paper is available online. drug histories and data from hospital notes were collected. ► Only patients who did not die as a result of the To view these files please visit Participants Two expert cardiologists reviewed data the journal online (http:// dx. doi. proarrhythmia could be included. for 293 referred cases: 130 were included. Inclusion org/ 10. 1136/ bmjopen- 2017- ► Referral of cases by cardiologists alone may have criteria were new onset or exacerbation of pre-existing 016627). -

Atrial Fibrillation (ATRIA) Study
European Journal of Human Genetics (2014) 22, 297–306 & 2014 Macmillan Publishers Limited All rights reserved 1018-4813/14 www.nature.com/ejhg REVIEW Atrial fibrillation: the role of common and rare genetic variants Morten S Olesen*,1,2,4, Morten W Nielsen1,2,4, Stig Haunsø1,2,3 and Jesper H Svendsen1,2,3 Atrial fibrillation (AF) is the most common cardiac arrhythmia affecting 1–2% of the general population. A number of studies have demonstrated that AF, and in particular lone AF, has a substantial genetic component. Monogenic mutations in lone and familial AF, although rare, have been recognized for many years. Presently, mutations in 25 genes have been associated with AF. However, the complexity of monogenic AF is illustrated by the recent finding that both gain- and loss-of-function mutations in the same gene can cause AF. Genome-wide association studies (GWAS) have indicated that common single-nucleotide polymorphisms (SNPs) have a role in the development of AF. Following the first GWAS discovering the association between PITX2 and AF, several new GWAS reports have identified SNPs associated with susceptibility of AF. To date, nine SNPs have been associated with AF. The exact biological pathways involving these SNPs and the development of AF are now starting to be elucidated. Since the first GWAS, the number of papers concerning the genetic basis of AF has increased drastically and the majority of these papers are for the first time included in a review. In this review, we discuss the genetic basis of AF and the role of both common and rare genetic variants in the susceptibility of developing AF. -

Sudden Cardiac Death in Heart Failure: What Do We Need to Know in 2018 ?
Sudden Cardiac Death in Heart Failure: What do we need to know in 2018 ? Juan M. Aranda, Jr. MD FACC Professor of Medicine Director of Heart Failure and Cardiac Transplantation University of Florida Disclosures Consultant for Zoll LifeVest. 1 Sudden Cardiac Death Statistics • One of the most common causes of death in developed countries: Incidence Survival (cases/year) Worldwide 3,000,000 1 <1% U.S. 450,000 2 5% W. Europe 400,000 3 <5% • High recurrence rate 1 Myerberg RJ, Catellanos A. Cardiac Arrest and Sudden Cardiac Death. In: Braunwald E, ed. Heart Disease: A Textbook of Cardiovascular Medicine . 5 th Ed. New York: WB Saunders. 1997: 742-779. 2 Circulation. 2001; 104: 2158-2163. 3 Vreede-Swagemakers JJ et al. J Am Coll Cardiol 1997; 30: 1500-1505. Leading cause of Death in the US Septicemia SCA is a leading cause of Nephritis death in the U.S., second to Alzheimer’s Disease all cancers combined . Influenza/Pneumonia Diabetes Accidents/Injuries Chronic Lower Respiratory Diseases Cerebrovascular Disease Other Cardiac Causes Sudden Cardiac Arrest (SCA) All Cancers 0% 5% 10% 15% 20% 25% National Vital Statistics Report. 2001;49;11. MMWR. 2002;51:123-126. 2 Disease States Associated with SCD 1) Atherosclerotic CAD 2) Dilated Cardiomyophay: 10% of SCD cases in adults. 3) Hypertrophic Cardiomyopathy: 2/1,000 young adults. 48% of SCD in athletes ≤ 35yo. 4) Valvular Heart Disease 5) Congenital Heart Disease: Four conditions associated with increased post-op risk of SCD (Tetrology of Fallot, transposition of the great vessels, Aortic Stenosis, pulmonary vascular obstruction). -

Should Genetic Testing Be Recommended for Long QT Syndrome Patients and Their Relatives?
The Science Journal of the Lander College of Arts and Sciences Volume 11 Number 1 Fall 2017 - 2017 Should Genetic Testing Be Recommended for Long QT Syndrome Patients and Their Relatives? Menachem Braun Touro College Follow this and additional works at: https://touroscholar.touro.edu/sjlcas Part of the Cardiovascular Diseases Commons, and the Medical Genetics Commons Recommended Citation Braun, M. (2017). Should Genetic Testing Be Recommended for Long QT Syndrome Patients and Their Relatives?. The Science Journal of the Lander College of Arts and Sciences, 11(1). Retrieved from https://touroscholar.touro.edu/sjlcas/vol11/iss1/8 This Article is brought to you for free and open access by the Lander College of Arts and Sciences at Touro Scholar. It has been accepted for inclusion in The Science Journal of the Lander College of Arts and Sciences by an authorized editor of Touro Scholar. For more information, please contact [email protected]. Should Genetic Testing be Recommended for Long QT Syndrome Patients and Their Relatives? Menachem Braun Menachem Braun graduated with a BS in Biology in September 2017. Abstract 7KH/RQJ476\QGURPH /476 LVDIDPLOLDOSRWHQWLDOO\IDWDOFDUGLDFDUUK\WKPLD7UDGLWLRQDOO\LWKDVEHHQGLDJQRVHGE\(&* Molecular studies have provided evidence that LQTS can be caused by a range of underlying molecular abnormalities. Genetic research has proven that different forms of LQTS have different genotypic bases. Therefore, it has become possible to diagnose WKHVSHFLÀFW\SHRIGLVHDVHJHQHWLFDOO\7KLVVWXG\H[DPLQHVWKHDGYDQFHPHQWVPDGHLQWKHSDVWWKLUW\\HDUVLQXQGHUVWDQGLQJ /476DQGUHVHDUFKUHJDUGLQJWKHXVHRIJHQHWLFWHVWLQJLQRUGHUWRGHWHUPLQHWKHEHQHÀWVRIJHQHWLFWHVWLQJIRUWKLVGLVHDVH$ survey of original studies which produced the information is presented here, and provides the reader with an understanding of the PHFKDQLFVRIWKHGLVHDVHDQGKRZWKH\GLIIHULQWKHVHYHUDOJHQHWLFYDULDQWV5HVHDUFKVKRZVWKDWWKHEHQHÀWRIJHQHWLFWHVWLQJ must be weighed against the personal implications in may have for a particular patient and his or her family. -

Latest Diagnostic and Treatment Strategies for the Congenital Long QT Syndromes
Latest Diagnostic and Treatment Strategies for the Congenital Long QT Syndromes Michael J. Ackerman, MD, PhD Windland Smith Rice Cardiovascular Genomics Research Professor Professor of Medicine, Pediatrics, and Pharmacology Director, Long QT Syndrome Clinic and the Mayo Clinic Windland Smith Rice Sudden Death Genomics Laboratory President, Sudden Arrhythmia Death Syndromes (SADS) Foundation Learning Objectives to Disclose: • To RECOGNIZE the “faces” (phenotypes) of the congenital long QT syndromes (LQTS) • To CRITIQUE the various diagnostic modalities used in the evaluation of LQTS and UNDERSTAND their limitations • To ASSESS the currently available treatment options for the various LQT syndromes and EVALUATE their efficacy WINDLAND Smith Rice Sudden Death Genomics Laboratory Conflicts of Interest to Disclose: • Consultant – Boston Scientific, Gilead Sciences, Medtronic, St. Jude Medical, and Transgenomic/FAMILION • Royalties – Transgenomic/FAMILION Congenital Long QT Syndrome Normal QT interval QT QT Prolonged QT 1. Syncope 2. Seizures 3. Sudden death Torsades de pointes Congenital Long QT Syndrome Normal QT interval QT QT ♥ 1957 – first clinical description – JLNS ♥ 1960s – RomanoProlonged-Ward QT syndrome ♥ 1983 – “Schwartz/Moss score”1. Syncope ♥ 1991 – first LQTS chromosome locus 2. Seizures ♥ March 10, 1995 – birth of cardiac 3. Sudden channelopathies death Torsades de pointes Congenital Long QT Syndrome Normal QT interval QT QT Prolonged QT 1. Syncope 2. Seizures 3. Sudden death Torsades de pointes Congenital Long QT Syndrome Normal -

Long QT Syndrome
orphananesthesia Anaesthesia recommendations for patients suffering from Long QT syndrome Disease name: Long QT syndrome ICD 10: I45.8 Synonyms: LQTS Related syndromes: Romano-Ward S., Jervell and Lange-Nielsen S., Timothy S. Long QT syndrome (LQTS) is a cardiac disorder resulting from malfunction of cardiac ion channels. It can be divided in congenital (c-LQTS) and acquired (a-LQTS) forms. Typical is a prolongation of the QT interval on ECG and a propensity to ventricular tachyarrhythmias which can lead to a characteristic polymorphic ventricular tachycardia known as “torsades de pointes”. There is a risk of syncope, cardiac arrest or sudden death. The diagnosis is based on ECG findings, clinical and family history. The Schwartz score defines the probability of LQTS according to three categories: 1 point, low probability of LQTS; (2) 2 or 3 points, intermediate probability of LQTS; and (3) ≥3.5 points, high probability of LQTS. THE TABLE OF THE SCHWARTZ SCORE should be added here: Table 2 of the paper Schwartz PJ and Crotti L. QTc behavior during exercise and genetic testing for the Long-QT Syndrome: Circulation 2011;124:2181-2184. Medicine in progress Perhaps new knowledge Every patient is unique Perhaps the diagnostic is wrong Find more information on the disease, its centres of reference and patient organisations on Orphanet: www.orpha.net 1 Typical surgery The typical surgery for LQTS is left cardiac sympathetic denervation, which is recommended in patients symptomatic for cardiac events despite beta-blocker (BB) therapy or in patients in whom BBs are contraindicated or not tolerated. For therapeutic reasons the implantation of a cardioverter defibrillator (ICD) is recommended in patients with a previous cardiac arrest or in patients with recurrent syncopal events despite antiadrenergic interventions. -

Cardiac Arrest Versus Heart Attack Flyer
VS. HEART ATTACK CARDIAC ARREST VS. HEART ATTACK People often use these terms interchangeably, but they are not the same. WHAT IS CARDIAC ARREST? WHAT IS A HEART ATTACK? CARDIAC ARREST occurs when A HEART ATTACK occurs when the heart malfunctions and blood flow to the heart is blocked. stops beating unexpectedly. A blocked artery prevents oxygen-rich Cardiac arrest is triggered by an blood from reaching a section of the heart. electrical malfunction in the heart that If the blocked artery is not reopened Cardiac arrest is A heart attack is quickly, the part of the heart normally causes an irregular heartbeat an “ELECTRICAL” a “CIRCULATION” (arrhythmia). With its pumping action nourished by that artery begins to die. disrupted, the heart cannot pump blood problem. problem. to the brain, lungs and other organs. WHAT HAPPENS Symptoms of a heart attack may be WHAT HAPPENS immediate and may include intense Block Atery Seconds later, a person becomes discomfort in the chest or other areas unresponsive, is not breathing of the upper body, shortness of or is only gasping. Death occurs breath, cold sweats, and/or nausea/ within minutes if the victim vomiting. More often, though, does not receive treatment. symptoms start slowly and persist for hours, days or weeks before a heart attack. Unlike with cardiac arrest, the WHAT TO DO heart usually does not stop beating during a heart attack. The longer the Cardiac arrest person goes without treatment, the can be reversible A greater the damage. in some victims if it’s treated within a few minutes. First, The heart attack symptoms in women can call your local emergency number be different than men (shortness of breath, and start CPR right away. -

Update on the Diagnosis and Management of Familial Long QT Syndrome
Heart, Lung and Circulation (2016) 25, 769–776 POSITION STATEMENT 1443-9506/04/$36.00 http://dx.doi.org/10.1016/j.hlc.2016.01.020 Update on the Diagnosis and Management of Familial Long QT Syndrome Kathryn E Waddell-Smith, FRACP a,b, Jonathan R Skinner, FRACP, FCSANZ, FHRS, MD a,b*, members of the CSANZ Genetics Council Writing Group aGreen Lane Paediatric and Congenital Cardiac Services, Starship Children’s Hospital, Auckland New Zealand bDepartment[5_TD$IF] of Paediatrics,[6_TD$IF] Child[7_TD$IF] and[8_TD$IF] Youth[9_TD$IF] Health,[10_TD$IF] University of Auckland, Auckland, New Zealand Received 17 December 2015; accepted 20 January 2016; online published-ahead-of-print 5 March 2016 This update was reviewed by the CSANZ Continuing Education and Recertification Committee and ratified by the CSANZ board in August 2015. Since the CSANZ 2011 guidelines, adjunctive clinical tests have proven useful in the diagnosis of LQTS and are discussed in this update. Understanding of the diagnostic and risk stratifying role of LQTS genetics is also discussed. At least 14 LQTS genes are now thought to be responsible for the disease. High-risk individuals may have multiple mutations, large gene rearrangements, C-loop mutations in KCNQ1, transmembrane mutations in KCNH2, or have certain gene modifiers present, particularly NOS1AP polymorphisms. In regards to treatment, nadolol is preferred, particularly for long QT type 2, and short acting metoprolol should not be used. Thoracoscopic left cardiac sympathectomy is valuable in those who cannot adhere to beta blocker therapy, particularly in long QT type 1. Indications for ICD therapies have been refined; and a primary indication for ICD in post-pubertal females with long QT type 2 and a very long QT interval is emerging. -

Andersen-Tawil Syndrome
Andersen-Tawil syndrome Description Andersen-Tawil syndrome is a disorder that causes episodes of muscle weakness ( periodic paralysis), changes in heart rhythm (arrhythmia), and developmental abnormalities. Periodic paralysis begins early in life, and episodes last from hours to days. These episodes may occur after exercise or long periods of rest, but they often have no obvious trigger. Muscle strength usually returns to normal between episodes. However, mild muscle weakness may eventually become permanent. In people with Andersen-Tawil syndrome, the most common changes affecting the heart are ventricular arrhythmia, which is a disruption in the rhythm of the heart's lower chambers (the ventricles), and long QT syndrome. Long QT syndrome is a heart condition that causes the heart (cardiac) muscle to take longer than usual to recharge between beats. The irregular heartbeats can lead to discomfort, such as the feeling that the heart is skipping beats (palpitations). Uncommonly, the irregular heartbeats can cause fainting (syncope), and even more rarely, sudden death. Physical abnormalities associated with Andersen-Tawil syndrome typically affect the face, other parts of the head, and the limbs. These features often include a very small lower jaw (micrognathia), dental abnormalities (such as crowded teeth), low-set ears, widely spaced eyes, fusion (syndactyly) of the second and third toes, and unusual curving of the fingers or toes (clinodactyly). Some affected people also have short stature and an abnormal side-to-side curvature of the spine (scoliosis). The signs and symptoms of Andersen-Tawil syndrome vary widely, and they can be different even among affected members of the same family.