Spatial and Temporal Expression of High Mobility Group Nucleosome-Binding (HMGN) Genes in Brain Areas Associated with Cognition in Individuals with Down Syndrome
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

University of California, San Diego
UNIVERSITY OF CALIFORNIA, SAN DIEGO The post-terminal differentiation fate of RNAs revealed by next-generation sequencing A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Biomedical Sciences by Gloria Kuo Lefkowitz Committee in Charge: Professor Benjamin D. Yu, Chair Professor Richard Gallo Professor Bruce A. Hamilton Professor Miles F. Wilkinson Professor Eugene Yeo 2012 Copyright Gloria Kuo Lefkowitz, 2012 All rights reserved. The Dissertation of Gloria Kuo Lefkowitz is approved, and it is acceptable in quality and form for publication on microfilm and electronically: __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ Chair University of California, San Diego 2012 iii DEDICATION Ma and Ba, for your early indulgence and support. Matt and James, for choosing more practical callings. Roy, my love, for patiently sharing the ups and downs of this journey. iv EPIGRAPH It is foolish to tear one's hair in grief, as though sorrow would be made less by baldness. ~Cicero v TABLE OF CONTENTS Signature Page .............................................................................................................. iii Dedication .................................................................................................................... -

Transcriptome Analyses of Rhesus Monkey Pre-Implantation Embryos Reveal A
Downloaded from genome.cshlp.org on September 23, 2021 - Published by Cold Spring Harbor Laboratory Press Transcriptome analyses of rhesus monkey pre-implantation embryos reveal a reduced capacity for DNA double strand break (DSB) repair in primate oocytes and early embryos Xinyi Wang 1,3,4,5*, Denghui Liu 2,4*, Dajian He 1,3,4,5, Shengbao Suo 2,4, Xian Xia 2,4, Xiechao He1,3,6, Jing-Dong J. Han2#, Ping Zheng1,3,6# Running title: reduced DNA DSB repair in monkey early embryos Affiliations: 1 State Key Laboratory of Genetic Resources and Evolution, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, Yunnan 650223, China 2 Key Laboratory of Computational Biology, CAS Center for Excellence in Molecular Cell Science, Collaborative Innovation Center for Genetics and Developmental Biology, Chinese Academy of Sciences-Max Planck Partner Institute for Computational Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai 200031, China 3 Yunnan Key Laboratory of Animal Reproduction, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, Yunnan 650223, China 4 University of Chinese Academy of Sciences, Beijing, China 5 Kunming College of Life Science, University of Chinese Academy of Sciences, Kunming, Yunnan 650204, China 6 Primate Research Center, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, 650223, China * Xinyi Wang and Denghui Liu contributed equally to this work 1 Downloaded from genome.cshlp.org on September 23, 2021 - Published by Cold Spring Harbor Laboratory Press # Correspondence: Jing-Dong J. Han, Email: [email protected]; Ping Zheng, Email: [email protected] Key words: rhesus monkey, pre-implantation embryo, DNA damage 2 Downloaded from genome.cshlp.org on September 23, 2021 - Published by Cold Spring Harbor Laboratory Press ABSTRACT Pre-implantation embryogenesis encompasses several critical events including genome reprogramming, zygotic genome activation (ZGA) and cell fate commitment. -

DNA Methylation Changes in Down Syndrome Derived Neural Ipscs Uncover Co-Dysregulation of ZNF and HOX3 Families of Transcription
Laan et al. Clinical Epigenetics (2020) 12:9 https://doi.org/10.1186/s13148-019-0803-1 RESEARCH Open Access DNA methylation changes in Down syndrome derived neural iPSCs uncover co- dysregulation of ZNF and HOX3 families of transcription factors Loora Laan1†, Joakim Klar1†, Maria Sobol1, Jan Hoeber1, Mansoureh Shahsavani2, Malin Kele2, Ambrin Fatima1, Muhammad Zakaria1, Göran Annerén1, Anna Falk2, Jens Schuster1 and Niklas Dahl1* Abstract Background: Down syndrome (DS) is characterized by neurodevelopmental abnormalities caused by partial or complete trisomy of human chromosome 21 (T21). Analysis of Down syndrome brain specimens has shown global epigenetic and transcriptional changes but their interplay during early neurogenesis remains largely unknown. We differentiated induced pluripotent stem cells (iPSCs) established from two DS patients with complete T21 and matched euploid donors into two distinct neural stages corresponding to early- and mid-gestational ages. Results: Using the Illumina Infinium 450K array, we assessed the DNA methylation pattern of known CpG regions and promoters across the genome in trisomic neural iPSC derivatives, and we identified a total of 500 stably and differentially methylated CpGs that were annotated to CpG islands of 151 genes. The genes were enriched within the DNA binding category, uncovering 37 factors of importance for transcriptional regulation and chromatin structure. In particular, we observed regional epigenetic changes of the transcription factor genes ZNF69, ZNF700 and ZNF763 as well as the HOXA3, HOXB3 and HOXD3 genes. A similar clustering of differential methylation was found in the CpG islands of the HIST1 genes suggesting effects on chromatin remodeling. Conclusions: The study shows that early established differential methylation in neural iPSC derivatives with T21 are associated with a set of genes relevant for DS brain development, providing a novel framework for further studies on epigenetic changes and transcriptional dysregulation during T21 neurogenesis. -

Download Download
Supplementary Figure S1. Results of flow cytometry analysis, performed to estimate CD34 positivity, after immunomagnetic separation in two different experiments. As monoclonal antibody for labeling the sample, the fluorescein isothiocyanate (FITC)- conjugated mouse anti-human CD34 MoAb (Mylteni) was used. Briefly, cell samples were incubated in the presence of the indicated MoAbs, at the proper dilution, in PBS containing 5% FCS and 1% Fc receptor (FcR) blocking reagent (Miltenyi) for 30 min at 4 C. Cells were then washed twice, resuspended with PBS and analyzed by a Coulter Epics XL (Coulter Electronics Inc., Hialeah, FL, USA) flow cytometer. only use Non-commercial 1 Supplementary Table S1. Complete list of the datasets used in this study and their sources. GEO Total samples Geo selected GEO accession of used Platform Reference series in series samples samples GSM142565 GSM142566 GSM142567 GSM142568 GSE6146 HG-U133A 14 8 - GSM142569 GSM142571 GSM142572 GSM142574 GSM51391 GSM51392 GSE2666 HG-U133A 36 4 1 GSM51393 GSM51394 only GSM321583 GSE12803 HG-U133A 20 3 GSM321584 2 GSM321585 use Promyelocytes_1 Promyelocytes_2 Promyelocytes_3 Promyelocytes_4 HG-U133A 8 8 3 GSE64282 Promyelocytes_5 Promyelocytes_6 Promyelocytes_7 Promyelocytes_8 Non-commercial 2 Supplementary Table S2. Chromosomal regions up-regulated in CD34+ samples as identified by the LAP procedure with the two-class statistics coded in the PREDA R package and an FDR threshold of 0.5. Functional enrichment analysis has been performed using DAVID (http://david.abcc.ncifcrf.gov/) -

Integrating Single-Step GWAS and Bipartite Networks Reconstruction Provides Novel Insights Into Yearling Weight and Carcass Traits in Hanwoo Beef Cattle
animals Article Integrating Single-Step GWAS and Bipartite Networks Reconstruction Provides Novel Insights into Yearling Weight and Carcass Traits in Hanwoo Beef Cattle Masoumeh Naserkheil 1 , Abolfazl Bahrami 1 , Deukhwan Lee 2,* and Hossein Mehrban 3 1 Department of Animal Science, University College of Agriculture and Natural Resources, University of Tehran, Karaj 77871-31587, Iran; [email protected] (M.N.); [email protected] (A.B.) 2 Department of Animal Life and Environment Sciences, Hankyong National University, Jungang-ro 327, Anseong-si, Gyeonggi-do 17579, Korea 3 Department of Animal Science, Shahrekord University, Shahrekord 88186-34141, Iran; [email protected] * Correspondence: [email protected]; Tel.: +82-31-670-5091 Received: 25 August 2020; Accepted: 6 October 2020; Published: 9 October 2020 Simple Summary: Hanwoo is an indigenous cattle breed in Korea and popular for meat production owing to its rapid growth and high-quality meat. Its yearling weight and carcass traits (backfat thickness, carcass weight, eye muscle area, and marbling score) are economically important for the selection of young and proven bulls. In recent decades, the advent of high throughput genotyping technologies has made it possible to perform genome-wide association studies (GWAS) for the detection of genomic regions associated with traits of economic interest in different species. In this study, we conducted a weighted single-step genome-wide association study which combines all genotypes, phenotypes and pedigree data in one step (ssGBLUP). It allows for the use of all SNPs simultaneously along with all phenotypes from genotyped and ungenotyped animals. Our results revealed 33 relevant genomic regions related to the traits of interest. -

Identification of Reproduction-Related Gene Polymorphisms Using Whole
INVESTIGATION Identification of Reproduction-Related Gene Polymorphisms Using Whole Transcriptome Sequencing in the Large White Pig Population Daniel Fischer,* Asta Laiho,† Attila Gyenesei,‡ and Anu Sironen*,1 *Natural Resources Institute Finland (Luke), Green Technology, Animal and Plant Genomics and Breeding, FI-31600 Jokioinen, Finland, †The Finnish Microarray and Sequencing Centre, Turku Centre for Biotechnology, University of Turku and Åbo Akademi University, Tykistökatu 6, FI-20520 Turku, Finland, and ‡Campus Science Support Facilities, Vienna Biocenter, A-1030 Vienna, Austria ORCID ID: 0000-0003-2064-6960 (D.F.) ABSTRACT Recent developments in high-throughput sequencing techniques have enabled large-scale KEYWORDS analysis of genetic variations and gene expression in different tissues and species, but gene expression oviduct patterns and genetic variations in livestock are not well-characterized. In this study, we have used high- testis throughput transcriptomic sequencing of the Finnish Large White to identify gene expression patterns and gene expression coding polymorphisms within the breed in the testis and oviduct. The main objective of this study was to polymorphism identify polymorphisms within genes that are highly and specifically expressed in male and/or female SNP reproductive organs. The differential expression (DE) analysis underlined 1234 genes highly expressed in pig the testis and 1501 in the oviduct. Furthermore, we used a novel in-house R-package hoardeR for the transcriptome identification of novel genes and their orthologs, which underlined 55 additional DE genes based on RNAseq orthologs in the human, cow, and sheep. Identification of polymorphisms in the dataset resulted in a total of reproduction 29,973 variants, of which 10,704 were known coding variants. -

Multi-Sample Full-Length Transcriptome Analysis of 22 Breast Cancer Clinical
bioRxiv preprint doi: https://doi.org/10.1101/2020.07.15.199851; this version posted July 16, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Multi-sample Full-length Transcriptome Analysis of 22 Breast Cancer Clinical Specimens with Long-Read Sequencing Shinichi Namba1, §, Toshihide Ueno1, Shinya Kojima1, Yosuke Tanaka1, Satoshi Inoue1, Fumishi Kishigami1, Noriko Maeda2, Tomoko Ogawa3, Shoichi Hazama4, Yuichi Shiraishi5, Hiroyuki Mano1, and Masahito Kawazu1* Author affiliations: 1Division of Cellular Signaling, 5Division of Genome Analysis Platform Development, Research Institute, National Cancer Center, Tokyo 104-0045, Japan; 2Department of Gastroenterological, Breast and Endocrine Surgery, 4Department of Translational Research and Developmental Therapeutics against Cancer, Yamaguchi University Graduate School of Medicine, Yamaguchi 755-8505, Japan; 3Department of Breast Surgery, Mie University Hospital, Mie 514-8507, Japan; § Present address, Department of Statistical Genetics, Osaka University Graduate School of Medicine, Osaka 565-0871, Japan; * Corresponding author E-mail:[email protected] (MK) bioRxiv preprint doi: https://doi.org/10.1101/2020.07.15.199851; this version posted July 16, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Abstract Although transcriptome alteration is considered as one of the essential drivers of carcinogenesis, conventional short-read RNAseq technology has limited researchers from directly exploring full-length transcripts, only focusing on individual splice sites. -
![A Genomic Atlas of Human Adrenal and Gonad Development [Version 2; Referees: 4 Approved] Ignacio Del Valle1, Federica Buonocore1, Andrew J](https://docslib.b-cdn.net/cover/1314/a-genomic-atlas-of-human-adrenal-and-gonad-development-version-2-referees-4-approved-ignacio-del-valle1-federica-buonocore1-andrew-j-711314.webp)
A Genomic Atlas of Human Adrenal and Gonad Development [Version 2; Referees: 4 Approved] Ignacio Del Valle1, Federica Buonocore1, Andrew J
Wellcome Open Research 2017, 2:25 Last updated: 08 NOV 2017 RESEARCH ARTICLE A genomic atlas of human adrenal and gonad development [version 2; referees: 4 approved] Ignacio del Valle1, Federica Buonocore1, Andrew J. Duncan1, Lin Lin1, Martino Barenco2, Rahul Parnaik1, Sonia Shah3,4, Mike Hubank5, Dianne Gerrelli2, John C. Achermann 1 1Genetics and Genomic Medicine, UCL Great Ormond Street Institute of Child Health, London, UK 2Developmental Biology and Cancer, UCL Great Ormond Street Institute of Child Health, London, UK 3Institute for Molecular Bioscience, University of Queensland, Brisbane, Australia 4Institute of Cardiovascular Science, University College London, London, UK 5The Centre for Molecular Pathology, Royal Marsden Hospital, Sutton, UK v2 First published: 07 Apr 2017, 2:25 (doi: 10.12688/wellcomeopenres.11253.1) Open Peer Review Latest published: 23 Oct 2017, 2:25 (doi: 10.12688/wellcomeopenres.11253.2) Referee Status: Abstract Background: In humans, the adrenal glands and gonads undergo distinct biological events between 6-10 weeks post conception (wpc), such as testis Invited Referees determination, the onset of steroidogenesis and primordial germ cell 1 2 3 4 development. However, relatively little is currently known about the genetic mechanisms underlying these processes. We therefore aimed to generate a detailed genomic atlas of adrenal and gonad development across these critical version 2 report report stages of human embryonic and fetal development. published Methods: RNA was extracted from 53 tissue samples between 6-10 wpc 23 Oct 2017 (adrenal, testis, ovary and control). Affymetrix array analysis was performed and differential gene expression was analysed using Bioconductor. A version 1 mathematical model was constructed to investigate time-series changes across published report report report report 07 Apr 2017 the dataset. -

A Yeast Phenomic Model for the Influence of Warburg Metabolism on Genetic Buffering of Doxorubicin Sean M
Santos and Hartman Cancer & Metabolism (2019) 7:9 https://doi.org/10.1186/s40170-019-0201-3 RESEARCH Open Access A yeast phenomic model for the influence of Warburg metabolism on genetic buffering of doxorubicin Sean M. Santos and John L. Hartman IV* Abstract Background: The influence of the Warburg phenomenon on chemotherapy response is unknown. Saccharomyces cerevisiae mimics the Warburg effect, repressing respiration in the presence of adequate glucose. Yeast phenomic experiments were conducted to assess potential influences of Warburg metabolism on gene-drug interaction underlying the cellular response to doxorubicin. Homologous genes from yeast phenomic and cancer pharmacogenomics data were analyzed to infer evolutionary conservation of gene-drug interaction and predict therapeutic relevance. Methods: Cell proliferation phenotypes (CPPs) of the yeast gene knockout/knockdown library were measured by quantitative high-throughput cell array phenotyping (Q-HTCP), treating with escalating doxorubicin concentrations under conditions of respiratory or glycolytic metabolism. Doxorubicin-gene interaction was quantified by departure of CPPs observed for the doxorubicin-treated mutant strain from that expected based on an interaction model. Recursive expectation-maximization clustering (REMc) and Gene Ontology (GO)-based analyses of interactions identified functional biological modules that differentially buffer or promote doxorubicin cytotoxicity with respect to Warburg metabolism. Yeast phenomic and cancer pharmacogenomics data were integrated to predict differential gene expression causally influencing doxorubicin anti-tumor efficacy. Results: Yeast compromised for genes functioning in chromatin organization, and several other cellular processes are more resistant to doxorubicin under glycolytic conditions. Thus, the Warburg transition appears to alleviate requirements for cellular functions that buffer doxorubicin cytotoxicity in a respiratory context. -

Genome-Wide DNA Methylation Analysis Reveals Molecular Subtypes of Pancreatic Cancer
www.impactjournals.com/oncotarget/ Oncotarget, 2017, Vol. 8, (No. 17), pp: 28990-29012 Research Paper Genome-wide DNA methylation analysis reveals molecular subtypes of pancreatic cancer Nitish Kumar Mishra1 and Chittibabu Guda1,2,3,4 1Department of Genetics, Cell Biology and Anatomy, University of Nebraska Medical Center, Omaha, NE, 68198, USA 2Bioinformatics and Systems Biology Core, University of Nebraska Medical Center, Omaha, NE, 68198, USA 3Department of Biochemistry and Molecular Biology, University of Nebraska Medical Center, Omaha, NE, 68198, USA 4Fred and Pamela Buffet Cancer Center, University of Nebraska Medical Center, Omaha, NE, 68198, USA Correspondence to: Chittibabu Guda, email: [email protected] Keywords: TCGA, pancreatic cancer, differential methylation, integrative analysis, molecular subtypes Received: October 20, 2016 Accepted: February 12, 2017 Published: March 07, 2017 Copyright: Mishra et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC-BY), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Pancreatic cancer (PC) is the fourth leading cause of cancer deaths in the United States with a five-year patient survival rate of only 6%. Early detection and treatment of this disease is hampered due to lack of reliable diagnostic and prognostic markers. Recent studies have shown that dynamic changes in the global DNA methylation and gene expression patterns play key roles in the PC development; hence, provide valuable insights for better understanding the initiation and progression of PC. In the current study, we used DNA methylation, gene expression, copy number, mutational and clinical data from pancreatic patients. -
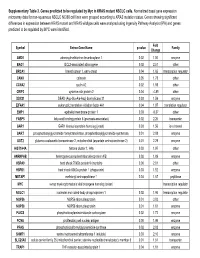
Supplementary Table 3. Genes Predicted to Be Regulated by Myc in KRAS Mutant NSCLC Cells
Supplementary Table 3. Genes predicted to be regulated by Myc in KRAS mutant NSCLC cells. Normalized basal gene expression microarray data for non-squamous NSCLC NCI60 cell lines were grouped according to KRAS mutation status. Genes showing significant differences in expression between KRAS mutant and KRAS wildtype cells were analyzed using Ingenuity Pathway Analysis (IPA) and genes predicted to be regulated by MYC were identified. Fold Symbol Entrez Gene Name p-value Family Change AMD1 adenosylmethionine decarboxylase 1 0.02 1.50 enzyme BAG1 BCL2-associated athanogene 0.02 2.57 other BRCA1 breast cancer 1, early onset 0.04 1.65 transcription regulator CANX calnexin 0.05 1.78 other CCNA2 cyclin A2 0.02 1.98 other CRIP2 cysteine-rich protein 2 0.04 -4.89 other DDX21 DEAD (Asp-Glu-Ala-Asp) box helicase 21 0.03 1.59 enzyme EIF4A1 eukaryotic translation initiation factor 4A1 0.04 1.87 translation regulator EMP1 epithelial membrane protein 1 0.03 -6.37 other FABP5 fatty acid binding protein 5 (psoriasis-associated) 0.02 2.26 transporter GAR1 GAR1 ribonucleoprotein homolog (yeast) 0.03 1.56 ion channel GART phosphoribosylglycinamide formyltransferase, phosphoribosylglycinamide synthetase, 0.01 2.08 enzyme GOT2 glutamic-oxaloacetic transaminase 2, mitochondrial (aspartate aminotransferase 2) 0.01 2.29 enzyme HIST1H4A histone cluster 1, H4a 0.03 1.97 other HNRNPAB heterogeneous nuclear ribonucleoprotein A/B 0.02 1.89 enzyme HSPA9 heat shock 70kDa protein 9 (mortalin) 0.00 2.01 other HSPD1 heat shock 60kDa protein 1 (chaperonin) 0.03 1.52 enzyme -

A Mutation in Histone H2B Represents a New Class of Oncogenic Driver
Author Manuscript Published OnlineFirst on July 23, 2019; DOI: 10.1158/2159-8290.CD-19-0393 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. A Mutation in Histone H2B Represents A New Class Of Oncogenic Driver Richard L. Bennett1, Aditya Bele1, Eliza C. Small2, Christine M. Will2, Behnam Nabet3, Jon A. Oyer2, Xiaoxiao Huang1,9, Rajarshi P. Ghosh4, Adrian T. Grzybowski5, Tao Yu6, Qiao Zhang7, Alberto Riva8, Tanmay P. Lele7, George C. Schatz9, Neil L. Kelleher9 Alexander J. Ruthenburg5, Jan Liphardt4 and Jonathan D. Licht1 * 1 Division of Hematology/Oncology, University of Florida Health Cancer Center, Gainesville, FL 2 Division of Hematology/Oncology, Northwestern University 3 Department of Cancer Biology, Dana Farber Cancer Institute and Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School 4 Department of Bioengineering, Stanford University 5 Department of Molecular Genetics and Cell Biology, The University of Chicago 6 Department of Chemistry, Tennessee Technological University 7 Department of Chemical Engineering, University of Florida 8 Bioinformatics Core, Interdisciplinary Center for Biotechnology Research, University of Florida 9 Department of Chemistry, Northwestern University, Evanston IL 60208 Running title: Histone mutations in cancer *Corresponding Author: Jonathan D. Licht, MD The University of Florida Health Cancer Center Cancer and Genetics Research Complex, Suite 145 2033 Mowry Road Gainesville, FL 32610 352-273-8143 [email protected] Disclosures: The authors have no conflicts of interest to declare Downloaded from cancerdiscovery.aacrjournals.org on September 27, 2021. © 2019 American Association for Cancer Research. Author Manuscript Published OnlineFirst on July 23, 2019; DOI: 10.1158/2159-8290.CD-19-0393 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited.