X-Ray Crystallographic Studies of the Metal Binding Sites on Recombinant Porcine Fructose-1,6-Bisphosphatase Jun-Yong Choe Iowa State University
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-
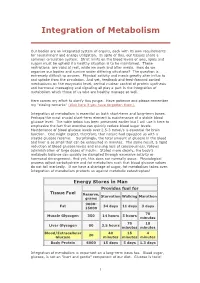
Integration of Metabolism
Integration of Metabolism Our bodies are an integrated system of organs, each with its own requirements for nourishment and energy utilization. In spite of this, our tissues share a common circulation system. Strict limits on the blood levels of ions, lipids and sugars must be upheld if a healthy situation is to be maintained. These restrictions are valid at rest, while we work and after meals. How do we organize our bodies and survive under differing situations? The question is extremely difficult to answer. Physical activity and meals greatly alter influx to and uptake from the circulation. And yet, feedback and feed-forward control mechanisms on the enzymatic level, central nuclear control of protein synthesis and hormonal messaging and signaling all play a part in the integration of metabolism which those of us who are healthy manage so well. Here comes my effort to clarify this jungle. Have patience and please remember my "closing remarks" (click here if you have forgotten them). Integration of metabolism is essential on both short-term and long-term bases. Perhaps the most crucial short-term element is maintenance of a stable blood glucose level. The table below has been presented earlier but I will use it here to emphasize the fact that exercise can quickly reduce blood sugar levels. Maintenance of blood glucose levels over 2.5-3 mmol/s is essential for brain function. One might expect, therefore, that nature had equipped us with a sizable glucose reserve. Surprisingly, the total amount of glucose in the blood and liver is so small that can be exhausted in minutes. -

Diagnostic Value of Serum Enzymes-A Review on Laboratory Investigations
Review Article ISSN 2250-0480 VOL 5/ ISSUE 4/OCT 2015 DIAGNOSTIC VALUE OF SERUM ENZYMES-A REVIEW ON LABORATORY INVESTIGATIONS. 1VIDYA SAGAR, M.SC., 2DR. VANDANA BERRY, MD AND DR.ROHIT J. CHAUDHARY, MD 1Vice Principal, Institute of Allied Health Sciences, Christian Medical College, Ludhiana 2Professor & Ex-Head of Microbiology Christian Medical College, Ludhiana 3Assistant Professor Department of Biochemistry Christian Medical College, Ludhiana ABSTRACT Enzymes are produced intracellularly, and released into the plasma and body fluids, where their activities can be measured by their abilities to accelerate the particular chemical reactions they catalyze. But different serum enzymes are raised when different tissues are damaged. So serum enzyme determination can be used both to detect cellular damage and to suggest its location in situ. Some of the biochemical markers such as alanine aminotransferase, aspartate aminotransferase, alkaline phasphatase, gamma glutamyl transferase, nucleotidase, ceruloplasmin, alpha fetoprotein, amylase, lipase, creatine phosphokinase and lactate dehydrogenase are mentioned to evaluate diseases of liver, pancreas, skeletal muscle, bone, etc. Such enzyme test may assist the physician in diagnosis and treatment. KEYWORDS: Liver Function tests, Serum Amylase, Lipase, CPK and LDH. INTRODUCTION mitochondrial AST is seen in extensive tissue necrosis during myocardial infarction and also in chronic Liver diseases like liver tissue degeneration DIAGNOSTIC SERUM ENZYME and necrosis². But lesser amounts are found in Enzymes are very helpful in the diagnosis of brain, pancreas and lung. Although GPT is plentiful cardiac, hepatic, pancreatic, muscular, skeltal and in the liver and occurs only in the small amount in malignant disorders. Serum for all enzyme tests the other tissues. -

GC-MS Metabolic Profile and -Glucosidase-, -Amylase-, Lipase-, and Acetylcholinesterase-Inhibitory Activities of Eight Peach
molecules Article GC-MS Metabolic Profile and α-Glucosidase-, α-Amylase-, Lipase-, and Acetylcholinesterase-Inhibitory Activities of Eight Peach Varieties Dasha Mihaylova 1,* , Ivelina Desseva 2,* , Aneta Popova 3 , Ivayla Dincheva 4 , Radka Vrancheva 2 , Anna Lante 5 and Albert Krastanov 1 1 Department of Biotechnology, Technological Faculty, University of Food Technologies, 4002 Plovdiv, Bulgaria; [email protected] 2 Department of Analytical Chemistry and Physical Chemistry, Technological Faculty, University of Food Technologies, 4002 Plovdiv, Bulgaria; [email protected] 3 Department of Catering and Tourism, Economics Faculty, University of Food Technologies, 4002 Plovdiv, Bulgaria; [email protected] 4 AgroBioInstitute, Agricultural Academy, 8 Dr. Tsankov Blvd., 1164 Sofia, Bulgaria; [email protected] 5 Department of Agronomy, Food, Natural Resources, Animals, and Environment—DAFNAE, Agripolis, University of Padova, 35020 Legnaro, Italy; [email protected] * Correspondence: [email protected] (D.M.); [email protected] (I.D.) Abstract: The inhibition of certain digestive enzymes by target food matrices represents a new approach in the treatment of socially significant diseases. Proving the ability of fruits to inhibit such enzymes can support the inclusion of specific varieties in the daily diets of patients with diabetes, obesity, Alzheimer’s disease, etc., providing them with much more than just valuable Citation: Mihaylova, D.; Desseva, I.; micro- and macromolecules. The current study aimed atidentifying and comparing the GC-MS Popova, A.; Dincheva, I.; Vrancheva, metabolic profiles of eight peach varieties (“Filina”, “Ufo 4, “Gergana”, “Laskava”, “July Lady”, R.; Lante, A.; Krastanov, A. GC-MS “Flat Queen”, “Evmolpiya”, and “Morsiani 90”) grown in Bulgaria (local and introduced) and to Metabolic Profile and α-Glucosidase-, evaluate the inhibitory potential of their extracts towards α-glucosidase, α-amylase, lipase, and α-Amylase-, Lipase-, and acetylcholinesterase. -

The Role of Phospholipase D (Pld) and Grb2 in Chemotaxis
THE ROLE OF PHOSPHOLIPASE D (PLD) AND GRB2 IN CHEMOTAXIS A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science By KATIE J. KNAPEK B.S., Indiana University of Pennyslvania, 2006 2008 Wright State University WRIGHT STATE UNIVERSITY SCHOOL OF GRADUATE STUDIES December 19, 2008 I HEREBY RECOMMEND THAT THE THESIS PREPARED UNDER MY SUPERVISON BY Katie J. Knapek ENTITLED The Role of Phospholipase D (PLD) and Grb2 in Chemotaxis BE ACCEPTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF Master of Science. _____________________________ Julian Gomez-Cambronero, Ph. D. Thesis Director _____________________________ Barbara Hull, Ph. D. Program Director Committee on Final Examination _______________________ Julian Gomez-Cambronero, Ph. D. _______________________ Nancy Bigley, Ph. D. _______________________ Mill Miller, Ph. D. ________________________ Joseph F. Thomas Jr., Ph. D. Dean, School of Graduate Studies ABSTRACT Knapek, Katie J. M.S., Department of Biological Sciences, Microbiology and Immunology Program, Wright State University, 2008. The Role of Phospholipase D (PLD) and Grb2 in Chemotaxis. Phospholipase D (PLD) is an enzyme that hydrolyzes phosphatidylcholine yielding choline and phosphatidic acid. PLD is activated by mitogens (lead to cell division) and motogens (leading to cell migration). PLD is known to contribute to cellular proliferation and deregulated expression of PLD has been implicated in several human cancers. PLD has been found to play a role in leukocyte chemotaxis and adhesion as studied through the formation of chemokine gradients. We have established a model of cell migration comprising three cell lines: macrophages RAW 264.7 and LR-5 (for innate defense), and fibroblast COS-7 cells (for wound healing). -

Multiple Roles of Phosphoinositide-Specific Phospholipase C Isozymes
BMB reports Mini Review Multiple roles of phosphoinositide-specific phospholipase C isozymes Pann-Ghill Suh1,*, Jae-Il Park1, Lucia Manzoli2, Lucio Cocco2, Joanna C. Peak3, Matilda Katan3, Kiyoko Fukami4, Tohru Kataoka5, Sanguk Yun1 & Sung Ho Ryu1 1Division of Molecular and Life Sciences, Pohang University of Science and Technology, Pohang, Korea, 2Cellular Signaling Laboratory, Department of Anatomical Sciences, University of Bologna, Via Irnerio, 48 I-40126, Bologna, Italy, 3Cancer Research UK Centre for Cell and Molecular Biology, Chester Beatty Laboratories, The Institute of Cancer Research, Fulham Road, London SW3 6JB, UK, 4Laboratory of Genome and Biosignal, Tokyo University of Pharmacy and Life Science, 1432-1 Horinouchi, Hachioji, 192-0392 Tokyo, Japan, 5Division of Molecular Biology, Department of Biochemistry and Molecular Biology, Kobe University Graduate School of Medicine, Chuo-ku, Kobe, Japan Phosphoinositide-specific phospholipase C is an effector mole- cellular calcium release (1-3; Fig. 1a). cule in the signal transduction process. It generates two sec- The first evidence of PLC activity was suggested by Hokin et ond messengers, inositol-1,4,5-trisphosphate and diacylglycer- al. in 1953 who reported specific hydrolysis of phospholipids ol from phosphatidylinositol 4,5-bisphosphate. Currently, thir- in pigeon’s pancreas slices after cholinergic stimulation (4). teen mammal PLC isozymes have been identified, and they are The authors showed that the enhanced turnover of phosphor- divided into six groups: PLC-β, -γ, -δ, -ε, -ζ and -η. Sequence ylinositol groups of phosphatidylinositol occurred in cells as a analysis studies demonstrated that each isozyme has more response to a variety of stimuli. In 1983, Streb et al. -

Pancreatic Amylase and Lipase Plasma Concentrations Are
Diabetes Care Volume 38, May 2015 e71 Pancreatic Amylase and Lipase Plasma David P. Sonne,1 Tina Vilsbøll,1 and Filip K. Knop1,2 Concentrations Are Unaffected by Increments in Endogenous GLP-1 Levels Following Liquid Meal Tests Diabetes Care 2015;38:e71–e72 | DOI: 10.2337/dc14-2751 Recent findings have suggested that amylase and lipase in plasma following patients with type 2 diabetes (P , incretin-based therapies promote pan- the ingestion of oral glucose and three 0.05), whereas lipase concentrations creatic inflammation and possibly cell isocaloric and isovolemic liquid mealsd were similar. Neither of the enzymes proliferation within the endocrine and all of which exerted normal endogenous increased following nutrient ingestion, exocrine pancreas (1). However, these GLP-1 secretion (4)din patients with suggesting that postprandial elevations studies have been met with substan- type 2 diabetes and matched control of endogenous GLP-1 (two- to three- tive criticism based on technical and subjects. fold) cannot trigger enzyme release methodical issues (2). Nevertheless, Detailed description of the experimen- from the human pancreas, at least not incretin-based therapies seem to result tal procedures and subjects was provided acutely. in small increases (within normal range) previously (4). In short, pancreas-specific These results suggest that the obser- in plasma concentrations of amylase amylase and lipase concentrations were vation of elevated plasma amylase and andlipaseinpatientsreceivingthese measured in plasma from 15 patients lipase -

Functional Role of Pentose Phosphate Pathway and Glutamine in Cancer Cell Metabolism Ibrahim Halil Polat
Functional role of pentose phosphate pathway and glutamine in cancer cell metabolism Ibrahim Halil Polat To cite this version: Ibrahim Halil Polat. Functional role of pentose phosphate pathway and glutamine in cancer cell metabolism. Human health and pathology. Universitat de Barcelona, 2016. English. NNT : 2016GREAS031. tel-01722703 HAL Id: tel-01722703 https://tel.archives-ouvertes.fr/tel-01722703 Submitted on 5 Mar 2018 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. THÈSE Pour obtenir le grade de DOCTEUR DE LA COMMUNAUTÉ UNIVERSITÉ GRENOBLE ALPES préparée dans le cadre d’une cotutelle entre la Communauté Université Grenoble Alpes et Université de Barcelona Spécialité : BIS-Biotechnologie, instrumentation, signal et imagerie pour la biologie, la médecine et l’environnement Arrêté ministériel : 25 mai 2016 Présentée par « Ibrahim Halil Polat » Thèse dirigée par « Philippe Sabatier » codirigée par « Marta Cascante » préparée au sein des Laboratoires « TIMC-IMAG » et « BQI » dans les Écoles Doctorales « EDISCE » et « Ecole Doctorale de l’Université de Barcelone » Rôle fonctionnel des pentoses phosphates et glutamine dans le métabolisme des cellules cancéreuses Thèse soutenue publiquement le « 4 novembre 2016 », devant le jury composé de : M. Santiago Imperial Rodenas Professeur à l’Université de Barcelone (Président) Mme Loranne Agius Professeur à l’Université de Newcastle Upon Tyne (Rapporteur) M. -

Unless Otherwise Noted, This Material Is Made Available
Author(s): Aken Desai, Michael Mathis, 2008 License: Unless otherwise noted, this material is made available under the terms of the Creative Commons Attribution – Share Alike 3.0 License: http://creativecommons.org/licenses/by-sa/3.0/ We have reviewed this material in accordance with U.S. Copyright Law and have tried to maximize your ability to use, share, and adapt it. Copyright holders of content included in this material should contact [email protected] with any quesCons, correcCons, or clarificaon regarding the use of content. For more informaon about how to cite these materials visit hHp://open.umich.edu/educaon/about/terms-of-use. Student works are presented as is and may be an interpretaon of faculty members’ lectures or assignments. These student works are not a product of faculty members. Faculty do not guarantee the accuracy of student work nor endorse them in any way. Any medical informaon in this material is intended to inform and educate and is not a tool for self-diagnosis or a replacement for medical evaluaon, advice, diagnosis or treatment by a healthcare professional. Please speak to your physician if you have quesCons about your medical condiCon. Viewer discreon is advised: Some medical content is graphic and may not be suitable for all viewers. Fatty Acids ctd. Tuesday, January 15, 2008 10:00 AM 14. What is the role of pentose pathway? Why is it important in fatty acid biosynthesis? What regulates the pentose pathway? How are pentoses formed without the oxidation of glucose‐6‐phosphate? a. Role is to provide NADPH and convert glucose into 5 carbon sugars for other pathways b. -

Lipase Interpretive Summary
Lipase Interpretive Summary Description: Lipase is an enzyme produced primarily in the pancreas to break down dietary fats in the small intestine. Decreased Lipase Common Causes Artifact o Hemolysis directly inhibits lipase activity in the assay Decreased lipase is not clinically significant. Increased Lipase Common Causes Pancreatic acinar cell damage o Inflammation o Necrosis o Neoplasia o Pancreatic duct obstruction Gastrointestinal inflammation Decreased renal clearance/inactivation (any condition that reduces glomerular filtration rate [GFR]) o Pre-renal conditions . Shock . Dehydration . Poor cardiac output o Renal disease o Post-renal disorders . Urinary tract obstruction or rupture Glucocorticoid treatment Uncommon Causes Hepatic neoplasia (dogs) Peritonitis Bowel obstruction Widespread steatitis Related Findings Pancreatic acinar cell damage o Increased amylase o Increased Spec cPL®/Spec fPL®. o Increased ALP +/- bilirubin o Increased glucose o Inflammatory leukogram on CBC Gastrointestinal inflammation o Increased BUN with gastric bleeding Decreased renal clearance/inactivation o Pre-renal . Increased hematocrit and total protein Generated by VetConnect® PLUS: Lipase Page 1 of 2 . Increased BUN, creatinine, and possibly phosphorous . Increased urine specific gravity . Decreased blood pressure (shock, poor cardiac output) o Renal . Increased BUN, creatinine, phosphorus . Isosthenuria o Post-renal . Increased BUN, creatinine, and possibly phosphorus . Abnormalities on urinalysis (blood, inflammation, crystals, etc.) . Abnormalities associated with urinary tract obstruction/rupture on abdominal ultrasound or contrast studies Additional Information Physiology Lipase is a cytoplasmic enzyme produced by the pancreas and released into the small intestine to catalyze the hydrolysis of triglycerides for absorption into the lacteals and gastrointestinal blood vessels. In addition to pancreatic acinar cells, lipase is produced by gastric mucosal cells, hepatocytes, and many extrahepatic cells (including adipocytes and myocytes). -
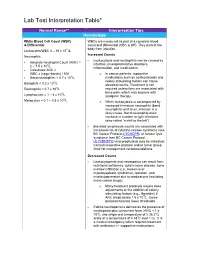
Lab Test Interpretation Table*
Lab Test Interpretation Table* Normal Range** Interpretation Tips Hematology White Blood Cell Count (WBC) WBCs are measured as part of a complete blood & Differential count and differential (CBC & diff). They protect the body from infection. Leukocytes/WBC 4 – 10 x 109 /L Increased Counts Neutrophils - Leukocytosis and neutrophilia can be caused by Absolute Neutrophil Count (ANC) = - infection, myeloproliferative disorders, 2 – 7.5 x 109/L inflammation, and medications. - Calculated ANC = WBC x (segs+bands) / 100 o In cancer patients, supportive - Band neutrophils: < 0.7 x 109/L medications such as corticosteroids and 9 colony stimulating factors can cause Basophils < 0.2 x 10 /L elevated counts. Treatment is not Eosinophils < 0.7 x 109/L required unless they are associated with 9 bone pain, which may improve with Lymphocytes = 1 – 4 x 10 /L analgesic therapy. 9 Monocytes = 0.1 – 0.8 x 10 /L o When leukocytosis is accompanied by increased immature neutrophils (band neutrophils) and fever, infection is a likely cause. Band neutrophils often increase in number to fight infections (also called “a shift to the left”). - Elevated lymphocyte counts are associated with increased risk of cytokine-release syndrome (see BC Cancer Protocol LYCHOPR) or tumour lysis syndrome (see BC Cancer Protocol ULYVENETO) and prophylaxis may be indicated. Consult respective protocol and/or tumor group chair for management recommendations. Decreased Counts - Leukocytopenia and neutropenia can result from nutritional deficiency, autoimmune disease, bone marrow infiltration (i.e., leukemia or myelodysplastic syndrome), radiation, and myelosuppression due to medications (including many cancer drugs). o Many treatment protocols require dose adjustments or the addition of colony stimulating factors (e.g., filgrastim) if ANC drops below 1.5 x 109/L. -

Are Common Polymorphisms of the Lipoprotein Lipase and Human Paraoxonase-1 Genes Associated with the Metabolic Syndrome in South
& Experim l e ca n i t in a l l C C f a Journal of Clinical & Experimental o r d Prakaschandra et al., J Clin Exp Cardiolog 2016, l i a o n l o r 7:6 g u y o J Cardiology DOI: 10.4172/2155-9880.1000452 ISSN: 2155-9880 Research Article Open Access Are Common Polymorphisms of the Lipoprotein Lipase and Human Paraoxonase-1 Genes Associated with the Metabolic Syndrome in South African Asian Indians? Rosaley Prakaschandra1, Michelle Gordon2 and Datshana P Naidoo3* 1Department of Biomedical and Clinical Technology, Durban University of Technology, South Africa 2School of Laboratory Medicine & Medical Sciences, University of KwaZulu-Natal, South Africa 3Department of Cardiology, University of KwaZulu-Natal, South Africa *Corresponding author: Datshana P Naidoo, Department of Cardiology, University of KwaZulu-Natal, Tel: 031-2402224/2207; E-mail: [email protected] Received date: May 16, 2016; Accepted date: June 20, 2016; Published date: June 30, 2016 Copyright: © 2016 Prakaschandra R, et al. This is an open -access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Abstract A cross-sectional study was performed to determine the possible contribution of the Human Paraoxonase-1 (PON1) and Lipoprotein Lipase (LPL) polymorphisms to the risk of the metabolic syndrome (MetS) in 817 participants of South African Asian Indian ancestry. Demographic and anthropometric data, including fasting blood for analysis of glycaemic and lipid parameters was collected. DNA was isolated from peripheral blood and allelic polymorphisms at positions Q192R, L55M in the PON1 gene and S447X and N291S in the LPL gene were studied using real-time PCR. -

The Role of Phosphatidylinositol-Specific Phospholipase-C in Plant Defense Signaling
The Role of Phosphatidylinositol-Specific Phospholipase-C in Plant Defense Signaling Ahmed M. Abd-El-Haliem Thesis committee Promotor Prof. Dr P.J.G.M. de Wit Professor of Phytopathology Wageningen University Co-promotor Dr M.H.A.J. Joosten Associate professor, Laboratory of Phytopathology Wageningen University Other members Prof. Dr H.J. Bouwmeester, Wageningen University Prof. Dr M.W. Prins, University of Amsterdam Prof. Dr G.C. Angenent, Wageningen University Dr S.H.E.J. Gabriёls, Monsanto Holland BV, Wageningen This research was conducted under the auspices of the Graduate School of Experimental Plant Sciences. The Role of Phosphatidylinositol-Specific Phospholipase-C in Plant Defense Signaling Ahmed M. Abd-El-Haliem Thesis submitted in fulfilment of the requirements for the degree of doctor at Wageningen University by the authority of the Rector Magnificus Prof. Dr M.J. Kropff, in the presence of the Thesis Committee appointed by the Academic Board to be defended in public on Thursday 23 October 2014 at 11.00 a.m. in the Aula. Ahmed M. Abd-El-Haliem The Role of Phosphatidylinositol-Specific Phospholipase-C in Plant Defense Signaling, 188 pages. PhD thesis, Wageningen University, Wageningen, NL (2014) With references, with summaries in Dutch and English ISBN 978-94-6257-118-1 TABLE OF CONTENTS CHAPTER 1 General Introduction & Thesis Outline 7 CHAPTER 2 Identification of Tomato Phosphatidylinositol-Specific 19 Phospholipase-C (PI-PLC) Family Members and the Role of PLC4 and PLC6 in HR and Disease Resistance CHAPTER 3 Defense Activation