The Role of Phospholipase D (Pld) and Grb2 in Chemotaxis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

ATP-Induced Focal Adhesion Kinase Activity Is Negatively Modulated by Phospholipase D2 in PC12 Cells
EXPERIMENTAL and MOLECULAR MEDICINE, Vol. 33, No. 3, 150-155, September 2001 ATP-induced focal adhesion kinase activity is negatively modulated by phospholipase D2 in PC12 cells Yoe-Sik Bae1 and Sung Ho Ryu1,2 Introduction 1 Division of Molecular and Life Sciences, Pohang University of Purinergic receptors have been reported to play impor- Science and Technology, Pohang 790-784, Korea tant roles on the regulation of neuronal cell functions 2 Corresponding author: Tel, +82-54-279-2292; (Communi et al., 2000; Di Iorio et al., 1998). ATP, a Fax, +82-54-279-2199; E-mail, [email protected] ligand for the receptors modulate various cellular re- sponses such as mitogenic and morphogenic activity in Accepted 18 September 2001 PC12 rat pheochromocytoma cells (Neary et al., 1996; Soltoff et al., 1998; Schindelholz et al., 2000). Stimu- Abbreviations: Fak, focal adhesion kinase; PLD, phospholipase D; lation of cells with ATP induces tyrosine phosphorylation PA, phosphatidic acid; PC, phosphatidylcholine; DAG, diacylglyc- of several cytoskeletal proteins and focal adhesion erol; PBt, phosphatidylbutanol; PKC, protein kinase C; PAP, phos- molecules such as focal adhesion kinase (Fak), proline- phatidic acid phosphohydrolase rich tyrosine kinase (Pyk2), and paxillin (Soltoff et al., 1998; Schindelholz et al., 2000). Since these cytosk- eleton-associated proteins have been regarded as important factors for the regulation of neuronal cell Abstract functions, the study on the regulatory mechanism for the proteins remains an important issue. Extracellular ATP has been known to modulate vari- Phospholipase D (PLD) catalyzes the hydrolysis of ous cellular responses including mitogenesis, secre- phosphatidylcholine (PC) into phosphatidic acid (PA) tion and morphogenic activity in neuronal cells. -

Revised Glossary for AQA GCSE Biology Student Book
Biology Glossary amino acids small molecules from which proteins are A built abiotic factor physical or non-living conditions amylase a digestive enzyme (carbohydrase) that that affect the distribution of a population in an breaks down starch ecosystem, such as light, temperature, soil pH anaerobic respiration respiration without using absorption the process by which soluble products oxygen of digestion move into the blood from the small intestine antibacterial chemicals chemicals produced by plants as a defence mechanism; the amount abstinence method of contraception whereby the produced will increase if the plant is under attack couple refrains from intercourse, particularly when an egg might be in the oviduct antibiotic e.g. penicillin; medicines that work inside the body to kill bacterial pathogens accommodation ability of the eyes to change focus antibody protein normally present in the body acid rain rain water which is made more acidic by or produced in response to an antigen, which it pollutant gases neutralises, thus producing an immune response active site the place on an enzyme where the antimicrobial resistance (AMR) an increasing substrate molecule binds problem in the twenty-first century whereby active transport in active transport, cells use energy bacteria have evolved to develop resistance against to transport substances through cell membranes antibiotics due to their overuse against a concentration gradient antiretroviral drugs drugs used to treat HIV adaptation features that organisms have to help infections; they -

NADPH-Dependent A-Oxidation of Unsaturated Fatty Acids With
Proc. Natl. Acad. Sci. USA Vol. 89, pp. 6673-6677, August 1992 Biochemistry NADPH-dependent a-oxidation of unsaturated fatty acids with double bonds extending from odd-numbered carbon atoms (5-enoyl-CoA/A3,A2-enoyl-CoA isomerase/A3',52'4-dienoyl-CoA isomerase/2,4-dienoyl-CoA reductase) TOR E. SMELAND*, MOHAMED NADA*, DEAN CUEBASt, AND HORST SCHULZ* *Department of Chemistry, City College of the City University of New York, New York, NY 10031; and tJoined Departments of Chemistry, Manhattan College/College of Mount Saint Vincent, Riverdale, NY 10471 Communicated by Salih J. Wakil, April 13, 1992 ABSTRACT The mitochondrial metabolism of 5-enoyl- a recent observation of Tserng and Jin (5) who reported that CoAs, which are formed during the (3-oxidation of unsaturated the mitochondrial -oxidation of 5-enoyl-CoAs is dependent fatty acids with double bonds extending from odd-numbered on NADPH. Their analysis of metabolites by gas chroma- carbon atoms, was studied with mitochondrial extracts and tography/mass spectrometry led them to propose that the purified enzymes of (3-oxidation. Metabolites were identified double bond of 5-enoyl-CoAs is reduced by NADPH to yield spectrophotometrically and by high performance liquid chro- the corresponding saturated fatty acyl-CoAs, which are then matography. 5-cis-Octenoyl-CoA, a putative metabolite of further degraded by P-oxidation. linolenic acid, was efficiently dehydrogenated by medium- This report addresses the question of how 5-enoyl-CoAs chain acyl-CoA dehydrogenase (EC 1.3.99.3) to 2-trans-5-cis- are chain-shortened by P-oxidation. We demonstrate that octadienoyl-CoA, which was isomerized to 3,5-octadienoyl- 5-enoyl-CoAs, after dehydrogenation to 2,5-dienoyl-CoAs, CoA either by mitochondrial A3,A2-enoyl-CoA isomerase (EC can be isomerized to 2,4-dienoyl-CoAs, which are reduced by 5.3.3.8) or by peroxisomal trifunctional enzyme. -

PLCG1) Mutations in Sézary Syndrome
This electronic thesis or dissertation has been downloaded from the King’s Research Portal at https://kclpure.kcl.ac.uk/portal/ Functional interrogations of Phospholipase C Gamma 1 (PLCG1) mutations in Sézary Syndrome Patel, Varsha Maheshkumar Awarding institution: King's College London The copyright of this thesis rests with the author and no quotation from it or information derived from it may be published without proper acknowledgement. END USER LICENCE AGREEMENT Unless another licence is stated on the immediately following page this work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International licence. https://creativecommons.org/licenses/by-nc-nd/4.0/ You are free to copy, distribute and transmit the work Under the following conditions: Attribution: You must attribute the work in the manner specified by the author (but not in any way that suggests that they endorse you or your use of the work). Non Commercial: You may not use this work for commercial purposes. No Derivative Works - You may not alter, transform, or build upon this work. Any of these conditions can be waived if you receive permission from the author. Your fair dealings and other rights are in no way affected by the above. Take down policy If you believe that this document breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 11. Oct. 2021 Functional interrogations of Phospholipase C Gamma 1 (PLCG1) mutations in Sézary Syndrome Varsha Maheshkumar Patel Skin Tumour Unit, St John’s Institute of Dermatology, School of Basic and Medical Biosciences, King’s College London. -

(4,5) Bisphosphate-Phospholipase C Resynthesis Cycle: Pitps Bridge the ER-PM GAP
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by UCL Discovery Topological organisation of the phosphatidylinositol (4,5) bisphosphate-phospholipase C resynthesis cycle: PITPs bridge the ER-PM GAP Shamshad Cockcroft and Padinjat Raghu* Dept. of Neuroscience, Physiology and Pharmacology, Division of Biosciences, University College London, London WC1E 6JJ, UK; *National Centre for Biological Sciences, TIFR-GKVK Campus, Bellary Road, Bangalore 560065, India Address correspondence to: Shamshad Cockcroft, University College London UK; Phone: 0044-20-7679-6259; Email: [email protected] Abstract Phospholipase C (PLC) is a receptor-regulated enzyme that hydrolyses phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) at the plasma membrane (PM) triggering three biochemical consequences, the generation of soluble inositol 1,4,5-trisphosphate (IP3), membrane– associated diacylglycerol (DG) and the consumption of plasma membrane PI(4,5)P2. Each of these three signals triggers multiple molecular processes impacting key cellular properties. The activation of PLC also triggers a sequence of biochemical reactions, collectively referred to as the PI(4,5)P2 cycle that culminates in the resynthesis of this lipid. The biochemical intermediates of this cycle and the enzymes that mediate these reactions are topologically distributed across two membrane compartments, the PM and the endoplasmic reticulum (ER). At the plasma membrane, the DG formed during PLC activation is rapidly converted to phosphatidic acid (PA) that needs to be transported to the ER where the machinery for its conversion into PI is localised. Conversely, PI from the ER needs to be rapidly transferred to the plasma membrane where it can be phosphorylated by lipid kinases to regenerate PI(4,5)P2. -

Identification of Lithium-Regulated Genes in Cultured Lymphoblasts of Lithium Responsive Subjects with Bipolar Disorder
Neuropsychopharmacology (2004) 29, 799–804 & 2004 Nature Publishing Group All rights reserved 0893-133X/04 $25.00 www.neuropsychopharmacology.org Identification of Lithium-Regulated Genes in Cultured Lymphoblasts of Lithium Responsive Subjects with Bipolar Disorder 1 1 1 2 3 4 Xiujun Sun , L Trevor Young , Jun-Feng Wang , Paul Grof , Gustavo Turecki , Guy A Rouleau ,5 and Martin Alda* 1Department of Psychiatry, University of Toronto, Toronto, Canada; 2Department of Psychiatry, University of Ottawa, Ottawa, Canada; 3 4 Department of Psychiatry, McGill University, Montreal, Canada; Center for Research in Neuroscience, McGill University, Montreal, Canada; 5 Department of Psychiatry, Dalhousie University, Halifax, Canada Lithium, a common drug for the treatment of bipolar disorder (BD), requires chronic administration to prevent recurrences of the illness. The necessity for long-term treatment suggests that changes in genes expression are involved in the mechanism of its action. We studied effects of lithium on gene expression in lymphoblasts from BD patients, all excellent responders to lithium prophylaxis. Gene expression was analyzed using cDNA arrays that included a total of 2400 cDNAs. We found that chronic lithium treatment at a therapeutically relevant concentration decreased the expression of seven genes in lymphoblasts from lithium responders. Five of these candidate lithium- regulated genes, including alpha1B-adrenoceptor (a1B-AR), acetylcholine receptor protein alpha chain precursor (ACHR), cAMP- 0 0 dependent 3 ,5 -cyclic phosphodiesterase 4D (PDE4D), substance-P receptor (SPR), and ras-related protein RAB7, were verified by Northern blotting analysis in lithium responders. None of these genes were regulated by lithium in healthy control subjects. When we compared the expression of these five genes between bipolar subjects and healthy control subjects at baseline, prior to lithium administration, we found that a1B-AR gene expression was higher in bipolar subjects than in healthy control subjects. -

Chapter 7. "Coenzymes and Vitamins" Reading Assignment
Chapter 7. "Coenzymes and Vitamins" Reading Assignment: pp. 192-202, 207-208, 212-214 Problem Assignment: 3, 4, & 7 I. Introduction Many complex metabolic reactions cannot be carried out using only the chemical mechanisms available to the side-chains of the 20 standard amino acids. To perform these reactions, enzymes must rely on other chemical species known broadly as cofactors that bind to the active site and assist in the reaction mechanism. An enzyme lacking its cofactor is referred to as an apoenzyme whereas the enzyme with its cofactor is referred to as a holoenzyme. Cofactors are subdivided into essential ions and organic molecules known as coenzymes (Fig. 7.1). Essential ions, commonly metal ions, may participate in substrate binding or directly in the catalytic mechanism. Coenzymes typically act as group transfer agents, carrying electrons and chemical groups such as acyl groups, methyl groups, etc., depending on the coenzyme. Many of the coenzymes are derived from vitamins which are essential for metabolism, growth, and development. We will use this chapter to introduce all of the vitamins and coenzymes. In a few cases--NAD+, FAD, coenzyme A--the mechanisms of action will be covered. For the remainder of the water-soluble vitamins, discussion of function will be delayed until we encounter them in metabolism. We also will discuss the biochemistry of the fat-soluble vitamins here. II. Inorganic cation cofactors Many enzymes require metal cations for activity. Metal-activated enzymes require or are stimulated by cations such as K+, Ca2+, or Mg2+. Often the metal ion is not tightly bound and may even be carried into the active site attached to a substrate, as occurs in the case of kinases whose actual substrate is a magnesium-ATP complex. -

The Protein Phosphatase PP2A Plays Multiple Roles in Plant Development by Regulation of Vesicle Traffic—Facts and Questions
International Journal of Molecular Sciences Review The Protein Phosphatase PP2A Plays Multiple Roles in Plant Development by Regulation of Vesicle Traffic—Facts and Questions Csaba Máthé *, Márta M-Hamvas, Csongor Freytag and Tamás Garda Department of Botany, Faculty of Science and Technology, University of Debrecen, H-4032 Debrecen, Hungary; [email protected] (M.M.-H.); [email protected] (C.F.); [email protected] (T.G.) * Correspondence: [email protected] Abstract: The protein phosphatase PP2A is essential for the control of integrated eukaryotic cell functioning. Several cellular and developmental events, e.g., plant growth regulator (PGR) mediated signaling pathways are regulated by reversible phosphorylation of vesicle traffic proteins. Reviewing present knowledge on the relevant role of PP2A is timely. We discuss three aspects: (1) PP2A regulates microtubule-mediated vesicle delivery during cell plate assembly. PP2A dephosphorylates members of the microtubule associated protein family MAP65, promoting their binding to microtubules. Regulation of phosphatase activity leads to changes in microtubule organization, which affects vesicle traffic towards cell plate and vesicle fusion to build the new cell wall between dividing cells. (2) PP2A-mediated inhibition of target of rapamycin complex (TORC) dependent signaling pathways contributes to autophagy and this has possible connections to the brassinosteroid signaling pathway. (3) Transcytosis of vesicles transporting PIN auxin efflux carriers. PP2A regulates vesicle localization and recycling of PINs related to GNOM (a GTP–GDP exchange factor) mediated pathways. The proper intracellular traffic of PINs is essential for auxin distribution in the plant body, thus in whole Citation: Máthé, C.; M-Hamvas, M.; plant development. -

Enzymes Main Concepts: •Proteins Are Polymers Made up of Amino Acids
Biology 102 Karen Bledsoe, Instructor Notes http://www.wou.edu/~bledsoek/ Chapter 6, section 4 Topic: Enzymes Main concepts: • Proteins are polymers made up of amino acids. Enzymes are a category of proteins. • Enzymes are catalysts. They speed up the rate of a chemical reaction by reducing the activation energy, which is the energy needed to carry out the reaction. An example of a catalyst: if you put a lit match to a sugar cube, it’s hard to make the sugar catch fire. But if you rub a corner of the sugar cube with ash or charcoal, it catches more easily. The ash or charcoal acts as a catalyst. • Many important chemical reactions in living cells can happen spontaneously, but these reactions happen too rarely and too slowly to sustain life. Enzymes make the reactions happen more quickly and more often. • Some reactions happen too violently to support life. Sugar, when burned, released a lot of energy, but the energy is mostly heat. A rapid reaction like that would overheat our cells. Enzyme-controlled reactions can release energy from sugar in a way that captures most of the energy and loses less of the energy as heat, so that the energy is useful to the cell and does not overheat the cell. • The structure of enzymes allows them to carry out reactions. Each enzyme is shaped to carry out only one specific reaction. This is known as enzyme specificity. Amylase, for example, is an enzyme that breaks down starch into glucose. Amylase only breaks down starch; it does not break down any other molecule, nor does it build starch out of glucose. -

1. Introduction
1 1. Introduction 1.1 Phospholipids in cellular signalling Signalling through lipid metabolites has gained a lot of attention since the finding of MABEL R. and LOWELL E. HOKIN (1953) that [32P]-orthophosphate is incorporated into phosphatidylinositol upon acetylcholine stimulation. This reaction step is now recognised as part of the phosphoinositide cascade. Its activation results in the release of D-meso-inositol-1,4,5-trisphosphate and sn-1,2-diacylglycerol (DAG) from phosphatidyl- inositol-4,5-bisphosphate (PIP2) (BERRIDGE and IRVINE, 1984). These second messengers are products of a phospholipase C (PLC), other second messengers are generated by phospholi- pase A2 (PLA2) and phospholipase D (PLD) (Fig. 1). O O O- P R3 R1 O O O phospholipase D R2 O phospholipase C O phospholipase A2 Figure 1. Mechanism of action of phospholipases involved in signalling on phospholipids. R1 is mostly a saturated, R2 an unsaturated carbon chain and R3 is a polar head group like choline, ethanolamine or inositol. Activation of the phosphoinositide cascade occurs through many cell surface receptors and has been characterised in great detail. Its second messenger D-meso-inositol-1,4,5-tris- phosphate releases Ca2+ from intracellular stores whereas sn-1,2-diacylglycerol activates protein kinase C (PKC). At present, three PIP2-specific phospholipase C families are characte- rised, referred to as phospholipase Cb, phospholipase Cg and phospholipase Cd (REBECCHI and PENTYALA, 2000). Each of these families comprises several members and is used in different signal transduction pathways. Receptors coupled to heterotrimeric G-proteins activate phospholipase Cb through the latter while activation of phospholipase Cg is mediated via tyrosine kinase receptors. -
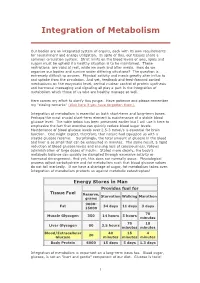
Integration of Metabolism
Integration of Metabolism Our bodies are an integrated system of organs, each with its own requirements for nourishment and energy utilization. In spite of this, our tissues share a common circulation system. Strict limits on the blood levels of ions, lipids and sugars must be upheld if a healthy situation is to be maintained. These restrictions are valid at rest, while we work and after meals. How do we organize our bodies and survive under differing situations? The question is extremely difficult to answer. Physical activity and meals greatly alter influx to and uptake from the circulation. And yet, feedback and feed-forward control mechanisms on the enzymatic level, central nuclear control of protein synthesis and hormonal messaging and signaling all play a part in the integration of metabolism which those of us who are healthy manage so well. Here comes my effort to clarify this jungle. Have patience and please remember my "closing remarks" (click here if you have forgotten them). Integration of metabolism is essential on both short-term and long-term bases. Perhaps the most crucial short-term element is maintenance of a stable blood glucose level. The table below has been presented earlier but I will use it here to emphasize the fact that exercise can quickly reduce blood sugar levels. Maintenance of blood glucose levels over 2.5-3 mmol/s is essential for brain function. One might expect, therefore, that nature had equipped us with a sizable glucose reserve. Surprisingly, the total amount of glucose in the blood and liver is so small that can be exhausted in minutes. -

Diagnostic Value of Serum Enzymes-A Review on Laboratory Investigations
Review Article ISSN 2250-0480 VOL 5/ ISSUE 4/OCT 2015 DIAGNOSTIC VALUE OF SERUM ENZYMES-A REVIEW ON LABORATORY INVESTIGATIONS. 1VIDYA SAGAR, M.SC., 2DR. VANDANA BERRY, MD AND DR.ROHIT J. CHAUDHARY, MD 1Vice Principal, Institute of Allied Health Sciences, Christian Medical College, Ludhiana 2Professor & Ex-Head of Microbiology Christian Medical College, Ludhiana 3Assistant Professor Department of Biochemistry Christian Medical College, Ludhiana ABSTRACT Enzymes are produced intracellularly, and released into the plasma and body fluids, where their activities can be measured by their abilities to accelerate the particular chemical reactions they catalyze. But different serum enzymes are raised when different tissues are damaged. So serum enzyme determination can be used both to detect cellular damage and to suggest its location in situ. Some of the biochemical markers such as alanine aminotransferase, aspartate aminotransferase, alkaline phasphatase, gamma glutamyl transferase, nucleotidase, ceruloplasmin, alpha fetoprotein, amylase, lipase, creatine phosphokinase and lactate dehydrogenase are mentioned to evaluate diseases of liver, pancreas, skeletal muscle, bone, etc. Such enzyme test may assist the physician in diagnosis and treatment. KEYWORDS: Liver Function tests, Serum Amylase, Lipase, CPK and LDH. INTRODUCTION mitochondrial AST is seen in extensive tissue necrosis during myocardial infarction and also in chronic Liver diseases like liver tissue degeneration DIAGNOSTIC SERUM ENZYME and necrosis². But lesser amounts are found in Enzymes are very helpful in the diagnosis of brain, pancreas and lung. Although GPT is plentiful cardiac, hepatic, pancreatic, muscular, skeltal and in the liver and occurs only in the small amount in malignant disorders. Serum for all enzyme tests the other tissues.