Analysis of the Coptis Chinensis Genome Reveals the Diversification
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Coptis Trifolia Conservation Assessment
CONSERVATION ASSESSMENT for Coptis trifolia (L.) Salisb. Originally issued as Management Recommendations December 1998 Marty Stein Reconfigured-January 2005 Tracy L. Fuentes USDA Forest Service Region 6 and USDI Bureau of Land Management, Oregon and Washington CONSERVATION ASSESSMENT FOR COPTIS TRIFOLIA Table of Contents Page List of Tables ................................................................................................................................. 2 List of Figures ................................................................................................................................ 2 Summary........................................................................................................................................ 4 I. NATURAL HISTORY............................................................................................................. 6 A. Taxonomy and Nomenclature.......................................................................................... 6 B. Species Description ........................................................................................................... 6 1. Morphology ................................................................................................................... 6 2. Reproductive Biology.................................................................................................... 7 3. Ecological Roles ............................................................................................................. 7 C. Range and Sites -

Medicinal Plants & Their Importance
Фомина Т.Н. MEDICINAL PLANTS & THEIR IMPORTANCE РОССИЙСКИЙ ГОСУДАРСТВЕННЫЙ АГРАРНЫЙ УНИВЕРСИТЕТ – МСХА имени К.А. ТИМИРЯЗЕВА [Введите название организации] Фомина Т.Н. 0 МОСКВА 2014 Т.Н. Фомина Английский язык в области производства и переработки лекарственных и эфиромасличных культур учебное пособие Рекомендовано к изданию учебно-методической комиссией факультета садоводства и ландшафтной архитектуры (протокол № 2 от 06 октября 2014 г.) ФГБОУ ВПО РГАУ-МСХА имени К.А. Тимирязева Издательство РГАУ-МСХА имени К.А. Тимирязева Москва 2014 1 УДК 802.0[663.8+665.52/.54](075.8) ББК 81.432.1:42.14я73 Ф 76 Фомина Т.Н. Английский язык в области производства и переработки лекарственных и эфиромасличных культур – Medicinal plants & their importance. Professional English for cultivation and processing technology of medicinal and essential-oil plants: учебное пособие/ Т.Н. Фомина. – М.: Изд-во РГАУ-МСХА имени К.А. Тимирязева, 2014. 138 с. Настоящее учебное пособие предназначено магистрам и аспирантам аграрного вуза, обучающихся по программе подготовки "Технологии производства продукции овощных и лекарственных растений" для направления 35.04.05 "Садоводство", а также студентам и аспирантам всех форм обучения агрономических специальностей и пограничных дисциплин сельскохозяйственных вузов и слушателям дополнительной образовательной программы «Переводчик в сфере профессиональной коммуникации». Рецензенты: Попченко М.И., к.б.н., доцент кафедры ботаники - РГАУ-МСХА имени К.А. Тимирязева Буковский С.Л., к. пед. н., ст. преподаватель кафедры иностранных языков - РГАУ-МСХА имени К.А. Тимирязева © Фомина Т.Н. © ФГБОУ ВПО РГАУ-МСХА имени К.А. Тимирязева, 2014 2 CONTENTS PAGE Introduction 4 Part I: Plants and people 6 Part II: Plants & their importance as alternative medicine 13 Part III: Classification of medicinal plants 25 Part IV: Plants list 37 Part V: Cultivation and management 53 Part VI: Obtaining raw material 62 Part VII: Industrial use 77 Part VIII: Essencial oils 89 Part IX: Standards and quality control. -
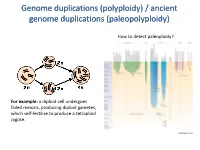
Polyploidy) / Ancient Genome Duplications (Paleopolyploidy
Genome duplications (polyploidy) / ancient genome duplications (paleopolyploidy) How to detect paleoploidy? For example: a diploid cell undergoes failed meiosis, producing diploid gametes, which self-fertilize to produce a tetraploid zygote. Timing of duplication by trees (phylogenetic timing) Phylogenetic timing of duplicates b Paramecium genome duplications Comparison of two scaffolds originating from a common ancestor at the recent WGD Saccharomyces cerevisiae Just before genome duplication Just after genome duplication More time after genome duplication Unaligned view (removing gaps just like in cerev has occurred) Saccharomyces cerevisiae Problem reciprocal gene loss (extreme case); how to solve? Problem reciprocal gene loss (extreme case); how to solve? Just before genome duplication Outgroup! Just after genome duplication Outgroup Just after genome duplication Outgroup More time after genome duplication Outgroup Problem (extreme case); how to solve? Outgroup Outgroup Outgroup Outgroup Outgroup Using other genomes Wong et al. 2002 PNAS Centromeres Vertebrate genome duplication Nature. 2011 Apr 10. [Epub ahead of print] Ancestral polyploidy in seed plants and angiosperms. Jiao Y, Wickett NJ, Ayyampalayam S, Chanderbali AS, Landherr L, Ralph PE, Tomsho LP, Hu Y, Liang H, Soltis PS, Soltis DE, Clifton SW, Schlarbaum SE, Schuster SC, Ma H, Leebens-Mack J, Depamphilis CW. Flowering plants Flowering MOSS Vertebrates Teleosts S. serevisiae and close relatives Paramecium Reconstructed map of genome duplications allows unprecedented mapping -

The Developmental and Genetic Bases of Apetaly in Bocconia Frutescens
Arango‑Ocampo et al. EvoDevo (2016) 7:16 DOI 10.1186/s13227-016-0054-6 EvoDevo RESEARCH Open Access The developmental and genetic bases of apetaly in Bocconia frutescens (Chelidonieae: Papaveraceae) Cristina Arango‑Ocampo1, Favio González2, Juan Fernando Alzate3 and Natalia Pabón‑Mora1* Abstract Background: Bocconia and Macleaya are the only genera of the poppy family (Papaveraceae) lacking petals; how‑ ever, the developmental and genetic processes underlying such evolutionary shift have not yet been studied. Results: We studied floral development in two species of petal-less poppies Bocconia frutescens and Macleaya cordata as well as in the closely related petal-bearing Stylophorum diphyllum. We generated a floral transcriptome of B. frutescens to identify MADS-box ABCE floral organ identity genes expressed during early floral development. We performed phylogenetic analyses of these genes across Ranunculales as well as RT-PCR and qRT-PCR to assess loci- specific expression patterns. We found that petal-to-stamen homeosis in petal-less poppies occurs through distinct developmental pathways. Transcriptomic analyses of B. frutescens floral buds showed that homologs of all MADS-box genes are expressed except for the APETALA3-3 ortholog. Species-specific duplications of other ABCE genes inB. frute- scens have resulted in functional copies with expanded expression patterns than those predicted by the model. Conclusions: Petal loss in B. frutescens is likely associated with the lack of expression of AP3-3 and an expanded expression of AGAMOUS. The genetic basis of petal identity is conserved in Ranunculaceae and Papaveraceae although they have different number of AP3 paralogs and exhibit dissimilar floral groundplans. -

A 'David and Goliath'
Current Genomics, 2002, 3, 563-576 563 Nucleolar Dominance: A ‘David and Goliath’ Chromatin Imprinting Process W. Viegas1,*, N. Neves1,2, M. Silva1, A. Caperta1,2, and L. Morais-Cecílio1 1Centro de Botânica Aplicada à Agricultura, Secção de Genética/DBEB, Instituto Superior de Agronomia, 1349-017 Lisboa, 2Departamento de Ciências Biológicas e Naturais, Universidade Lusófona de Humanidades e Tecnologias, Campo Grande 376, 1749-042 Lisboa, Portugal Abstract: Nucleolar dominance is an enigma. The puzzle of differential amphiplasty has remained unresolved since it was first recognised and described in Crepis hybrids by Navashin in 1934. Here we review the body of knowledge that has grown out of the many models that have tried to find the genetic basis for differential rRNA gene expression in hybrids, and present a new interpretation. We propose and discuss a chromatin imprinting model which re-interprets differential amphiplasty in terms of two genomes of differing size occupying a common space within the nucleus, and with heterochromatin as a key player in the scenario. Difference in size between two parental genomes induces an inherited epigenetic mark in the hybrid that allows patterns of chromatin organization to have positional effects on the neighbouring domains. This chromatin imprinting model can be also used to explain complex genomic interactions which transcend nucleolar dominance and which can account for the overall characteristics of hybrids. Gene expression in hybrids, relative to parentage, is seen as being based on the nuclear location of the sequences concerned within their genomic environment, and where the presence of particular repetitive DNA sequences are ‘sensed’, and render silent the adjacent information. -

Gene Loss and Adaptation in Saccharomyces Genomes
Genetics: Published Articles Ahead of Print, published on December 1, 2005 as 10.1534/genetics.105.048900 After the duplication: gene loss and adaptation in Saccharomyces genomes Paul F. Cliften*,1, Robert S. Fulton§, Richard K. Wilson*, §, and Mark Johnston* *Department of Genetics and §Genome Sequencing Center, Washington University School of Medicine, 660 South Euclid Ave., St. Louis, MO, 63110; 1Current address: Department of Biology, Utah State University, 5305 Old Main Hill, Logan, UT, 84322 Running head: Saccharomyces genomic duplications Key words: genomic duplication, comparative sequence analysis, Saccharomyces phylogeny, yeast Corresponding author: Mark Johnston Department of Genetics Campus Box 8232 Washington University Medical School 4566 Scott Ave. St. Louis, MO 63110 TEL: 314-362-2735 FAX: 314-362-2157 [email protected] ABSTRACT The ancient duplication of the Saccharomyces cerevisiae genome and subsequent massive loss of duplicated genes is apparent when it is compared to the genomes of related species that diverged before the duplication event. To learn more about the evolutionary effects of the duplication event, we compared the S. cerevisiae genome to other Saccharomyces genomes. We demonstrate that the whole genome duplication occurred before S. castellii diverged from S. cerevisiae. In addition to more accurately dating the duplication event, this finding allowed us to study the effects of the duplication on two separate lineages. Analyses of the duplication regions of the genomes indicate that most of the duplicated genes (approximately 85%) were lost before the speciation. Only a small amount of paralogous gene loss (4-6%) occurred after speciation. On the other hand, S. castellii appears to have lost several hundred genes that were not retained as duplicated paralogs. -

Study of Morphology and Outcome of Various Showing Measures With
Journal of Medicinal Plants Studies 2016; 4(5): 186-188 ISSN 2320-3862 JMPS 2016; 4(5): 186-188 Study of morphology and outcome of various © 2016 JMPS Received: 26-07-2016 showing measures with organic nutrient Accepted: 27-08-2016 application on newly introduced plant Coptis teeta Dhiman Mukherjee Directorate of Research, Bidhan (rare plant in earth) in Darjeeling hills Chandra Krishi Viswavidayalaya Kalyani, West Bengal, India Dhiman Mukherjee Abstract An experiment was carried out during the year of 2009 - 2012 at lava (2040 m asl), Darjeeling hill, under the aegis of Uttar Banga Krishi Viswavidyalaya, to evaluate the effect of various showing measures and organic nutrient application on newly introduced plant Coptis teeta in Darjeeling hill. Seedlings were planted in main field, to work out its morphology and other growth pattern with agronomic manipulation. Field experiment was conducted in split plot design with three replication, having four method of sowing in main plot, which includes viz. plain bed, sloppy land (terrace), furrow and undulated sowing, and four nutrient management practices in subplot viz. vermicompost (@0.50t/ha), FYM (@0 50 t/ha), leaf manure (@0 50 t/ha) and control (no nutrient). This is a small herb of 20-25 cm in hight. This has compound leaf, serrated into 3 part leaf blade ovate, triangular in shape. Root was fibrous in nature with small yellow coloured. Colour of the rhizome was golden yellow. Inflorescence includes 3-5 flowers, bract elliptic, 3 parted or pinnately divided, solitary, cymose. Rhizome weight was more found with sloppy land cultivation (3.51 g), and was at par with the ridge method of planting (2.96 g), and statistically better to rest of the imposed treatments of main plot. -

Isolation and Identification of Alkaloids from Macleaya Microcarpa by UHPLC–Q-TOF-MS and Their Cytotoxic Activity in Vitro, An
Sai et al. BMC Chemistry (2020) 14:5 https://doi.org/10.1186/s13065-020-0660-1 BMC Chemistry RESEARCH ARTICLE Open Access Isolation and identifcation of alkaloids from Macleaya microcarpa by UHPLC– Q-TOF-MS and their cytotoxic activity in vitro, antiangiogenic activity in vivo Chunmei Sai1,2, Jian’an Wang1, Binjie Li3, Lin Ding1, Huiyun Wang1, Qibao Wang1, Huiming Hua3, Fangpeng Zhang2 and Qiang Ren1* Abstract Background: Extensive bioactivities of alkaloids from the genus Macleaya (Macleaya cordata (Willd.) R. Br. and Macleaya microcarpa (Maxim.) Fedde) have been widely reported, as well as more and more concerned from the sci- entifc communities. However, systematic research on the phytochemical information of M. microcarpa is incomplete. The aim of this study was to rapidly and conveniently qualitative analyze alkaloids from M. microcarpa by ultra-perfor- mance liquid chromatography/quadrupole-time-of-fght mass spectrometry (UHPLC–Q-TOF-MS) using accurate mass weight and characteristic fragment ions, furthermore separate and identify the main alkaloids, test antitumor activity in vitro and antiangiogenic activity in vivo. Results: A total of 14 alkaloids from fruits of M. microcarpa were identifed by UHPLC–Q-TOF-MS, including 5 pro- topines, 2 benzophenanthridines, 1 dimer, 1 dihydrobenzophenanthridines and 5 unknown structure compounds. Two major alkaloids were isolated by various column chromatographic methods. Their structures were determined by NMR data and related literatures. The two major alkaloids were evaluated for intro cytotoxic activities against HL-60, MCF-7, A-549, and in vivo antiangiogenic activity using transgenic zebrafsh. Conclusions: Current qualitative method based on UHPLC–Q-TOF-MS technique provided a scientifc basis for isola- tion, structural identifcation, and in vitro or in vivo pharmacological further study of alkaloids from M. -

On Adulteration of Hydrastis Canadensis Root and Rhizome by Michael Tims, Phd Maryland University of Integrative Health, 7750 Montpelier Road, Laurel, MD 20723
on Adulteration of Hydrastis canadensis root and rhizome By Michael Tims, PhD Maryland University of Integrative Health, 7750 Montpelier Road, Laurel, MD 20723 Correspondence: e-mail Keywords: Hydrastis canadensis, goldenseal root, adulterant, adulteration Goal: The goal of this bulletin is to provide information and/or updates on issues regarding adulteration of goldenseal (Hydrastis canadensis) root to the international herbal industry and extended natural products community in general. It is intended to present the available data on occurrence of adulteration, the market situa- tion, and consequences for the consumer and the industry. 1 General Information 1.1 Common name: Goldenseal1,2 1.2 Other common names: English: Yellow root, yellow puccoon, ground raspberry, wild curcuma*, Indian dye, eye root, eye balm, Indian paint, jaundice root, Warnera3,4 French: Hydraste du Canada, hydraste, fard inolien, framboise de terre, sceau d’or5 German: Goldsiegelwurzel, Kanadische Gelbwurz, Kanadische Orangenwurzel5 Italian: Idraste, radice gialla6 Goldenseal Hydrastis canadensis Spanish: Hidrastis, hidrastis de Canadá, raíz de oro, scello de Photo ©2016 Steven Foster oro5 1.3 Accepted Latin binomial: Hydrastis canadensis7,8 1.4 Botanical family: Ranunculaceae 1.5 Plant part and form: Whole fresh or dry roots and rhizomes, powdered dry roots and rhizomes, hydroalcoholic and glycerin-water extracts and powdered dry extracts.9,10 Dried whole or powdered roots and rhizomes complying with the United States Pharmacopeia (USP) are required to contain -

Medicinal Plant Conservation
MEDICINAL Medicinal Plant PLANT SPECIALIST GROUP Conservation Silphion Volume 11 Newsletter of the Medicinal Plant Specialist Group of the IUCN Species Survival Commission Chaired by Danna J. Leaman Chair’s note . 2 Sustainable sourcing of Arnica montana in the International Standard for Sustainable Wild Col- Apuseni Mountains (Romania): A field project lection of Medicinal and Aromatic Plants – Wolfgang Kathe . 27 (ISSC-MAP) – Danna Leaman . 4 Rhodiola rosea L., from wild collection to field production – Bertalan Galambosi . 31 Regional File Conservation data sheet Ginseng – Dagmar Iracambi Medicinal Plants Project in Minas Gerais Lange . 35 (Brazil) and the International Standard for Sus- tainable Wild Collection of Medicinal and Aro- Conferences and Meetings matic Plants (ISSC-MAP) – Eleanor Coming up – Natalie Hofbauer. 38 Gallia & Karen Franz . 6 CITES News – Uwe Schippmann . 38 Conservation aspects of Aconitum species in the Himalayas with special reference to Uttaran- Recent Events chal (India) – Niranjan Chandra Shah . 9 Conservation Assessment and Management Prior- Promoting the cultivation of medicinal plants in itisation (CAMP) for wild medicinal plants of Uttaranchal, India – Ghayur Alam & Petra North-East India – D.K. Ved, G.A. Kinhal, K. van de Kop . 15 Ravikumar, R. Vijaya Sankar & K. Haridasan . 40 Taxon File Notices of Publication . 45 Trade in East African Aloes – Sara Oldfield . 19 Towards a standardization of biological sustain- List of Members. 48 ability: Wildcrafting Rhatany (Krameria lap- pacea) in Peru – Maximilian -

Berberine: Botanical Occurrence, Traditional Uses, Extraction Methods, and Relevance in Cardiovascular, Metabolic, Hepatic, and Renal Disorders
REVIEW published: 21 August 2018 doi: 10.3389/fphar.2018.00557 Berberine: Botanical Occurrence, Traditional Uses, Extraction Methods, and Relevance in Cardiovascular, Metabolic, Hepatic, and Renal Disorders Maria A. Neag 1, Andrei Mocan 2*, Javier Echeverría 3, Raluca M. Pop 1, Corina I. Bocsan 1, Gianina Cri¸san 2 and Anca D. Buzoianu 1 1 Department of Pharmacology, Toxicology and Clinical Pharmacology, “Iuliu Hatieganu” University of Medicine and Pharmacy, Cluj-Napoca, Romania, 2 Department of Pharmaceutical Botany, “Iuliu Hatieganu” University of Medicine and Pharmacy, Cluj-Napoca, Romania, 3 Department of Environmental Sciences, Universidad de Santiago de Chile, Santiago de Chile, Chile Edited by: Berberine-containing plants have been traditionally used in different parts of the world for Anna Karolina Kiss, the treatment of inflammatory disorders, skin diseases, wound healing, reducing fevers, Medical University of Warsaw, Poland affections of eyes, treatment of tumors, digestive and respiratory diseases, and microbial Reviewed by: Pinarosa Avato, pathologies. The physico-chemical properties of berberine contribute to the high diversity Università degli Studi di Bari Aldo of extraction and detection methods. Considering its particularities this review describes Moro, Italy various methods mentioned in the literature so far with reference to the most important Sylwia Zielinska, Wroclaw Medical University, Poland factors influencing berberine extraction. Further, the common separation and detection *Correspondence: methods like thin layer chromatography, high performance liquid chromatography, and Andrei Mocan mass spectrometry are discussed in order to give a complex overview of the existing [email protected] methods. Additionally, many clinical and experimental studies suggest that berberine Specialty section: has several pharmacological properties, such as immunomodulatory, antioxidative, This article was submitted to cardioprotective, hepatoprotective, and renoprotective effects. -

The First Comprehensive Phylogeny of Coptis (Ranunculaceae) and Its Implications for Character Evolution and Classification
RESEARCH ARTICLE The First Comprehensive Phylogeny of Coptis (Ranunculaceae) and Its Implications for Character Evolution and Classification Kun-Li Xiang1,2, Sheng-Dan Wu3, Sheng-Xian Yu1, Yang Liu4, Florian Jabbour5, Andrey S. Erst6,7, Liang Zhao8, Wei Wang1*, Zhi-Duan Chen1 1 State Key Laboratory of Systematic and Evolutionary Botany, Institute of Botany, Chinese Academy of Sciences, Beijing, 100093, China, 2 University of Chinese Academy of Sciences, Beijing, 100049, China, 3 College of Life Sciences, Shanxi Normal University, Linfen, 041004, China, 4 Department of Ecology and Evolutionary Biology, University of Connecticut, Storrs, Connecticut, 06269–3043, United States of America, 5 Muséum national d’Histoire naturelle, Institut de Systématique, Evolution, Biodiversité, UMR 7205 ISYEB MNHN/CNRS/UPMC/EPHE, Sorbonne Universités, Paris, 75005, France, 6 Central Siberian Botanical Garden of the Siberian Branch of Russian Academy of Sciences, Zolotodolinskaya str. 101, Novosibirsk, 630090, Russia, 7 Laboratory of Systematics and Phylogeny of Plants, National Research Tomsk State University, prospekt Lenina 36, Tomsk, 634050, Russia, 8 College of Life Sciences, Northwest A&F University, Yangling, Shaanxi, 712100, China * [email protected] OPEN ACCESS Citation: Xiang K-L, Wu S-D, Yu S-X, Liu Y, Jabbour F, Erst AS, et al. (2016) The First Comprehensive Abstract Phylogeny of Coptis (Ranunculaceae) and Its Implications for Character Evolution and Coptis (Ranunculaceae) contains 15 species and is one of the pharmaceutically most Classification. PLoS ONE 11(4): e0153127. important plant genera in eastern Asia. Understanding of the evolution of morphological doi:10.1371/journal.pone.0153127 characters and phylogenetic relationships within the genus is very limited.