Sulfur Dioxide Oxidation and Isotope Fractionation As a Function of Ph
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Crown Chemical Resistance Chart
Crown Polymers, Corp. 11111 Kiley Drive Huntley, IL. 60142 USA www.crownpolymers.com 847-659-0300 phone 847-659-0310 facisimile 888-732-1270 toll free Chemical Resistance Chart Crown Polymers Floor and Secondary Containment Systems Products: CrownShield covers the following five (4) formulas: CrownShield 50, Product No. 320 CrownCote, Product No. 401 CrownShield 40-2, Product No. 323 CrownShield 28, Product No. 322 CrownPro AcidShield, Product No. 350 CrownCote AcidShield, Product No. 430 CrownPro SolventShield, Product No. 351 CrownCote SolventShield, Product No. 440 This chart shows chemical resistance of Crown Polymers foundational floor and secondary containment product line that would be exposed to chemical spill or immersion conditions. The chart was designed to provide general product information. For specific applications, contact your local Crown Polymers Floor and Secondary Containment Representative or call direct to the factory. ; Resistant to chemical immersion up to 7 days followed by wash down with water 6 Spillage environments that will be cleaned up within 72 hours after initial exposure. 9 Not Recommended Chemical CrownShield SolventShield AcidShield Chemical CrownShield SolventShield AcidShield 1, 4-Dichloro-2-butene 9 6 6 Aluminum Bromate ; ; ; 1, 4-Dioxane 9 6 6 Aluminum Bromide ; ; ; 1-1-1 Trichloroethane 9 ; ; Aluminum Chloride ; ; ; 2, 4-Pentanedione 6 ; 6 Aluminum Fluoride (25%) ; ; ; 3, 4-Dichloro-1-butene 6 6 6 Aluminum Hydroxide ; ; ; 4-Picoline (0-50%) 9 6 6 Aluminum Iodine ; ; ; Acetic Acid (0-15%) 9 6 6 -

The Institute of Paper Chemistry
The Institute of Paper Chemistry Appleton, Wisconsin Doctor's Dissertation A Study of the Order and Nature of the Aspenwood Hemicellulose Removed During a Neutral Sulfite Semichemical Cook Robert Harold Quick June, 1955 A STUDY OF THE ORDER AND NATURE OF THE ASPENWOOD HEMICELLULOSE REMOVED DURING A NEUTRAL SULFITE SEMICHEMICAL COOK A thesis submitted by Robert Harold Quick B.S. in M.E. 1948, University of Washington M.S. 1953, Lawrence College in partial fulfillment of the requirements of The Institute of Paper Chemistry for the degree of Doctor of Philosophy from Lawrence College, Appleton, Wisconsin June, 1955 TABLE OF CONTENTS GLOSSARY iv INTRODUCTION Background of the Problem 1 Statement of the Problem 3 Historical Review 3 EXPERIMENTAL RESULTS Approach to the Problem 7 Wood Data 7 Cooking Conditions 7 Spent Liquor Study During a NSSC Cook Cook C2P1 8 Hydrolysis of C2P1 Spent Liquor 10 Approximate Organic Balance on C2P1SL 10 Study of the Pulp and Liquor Cooks C3P1, C4P2, and C5P1 17 Hydrolysis of Spent Liquor 19 Wood and Pulp Analyses 23 Spent Liquor "Hemicellulose" Analysis 31 DISCUSSION OF RESULTS Liquor Study 35 Pulp and Liquor Study 37 SUMMARY 51 CONCLUSIONS 54 APPENDIX I Experimental Procedures Preparation of Chips 56 Pulping 56 iii Preparation of Holocellulose 57 Extraction of Hemicellulose from Holocellulose 59 Recovery of "Hemicellulose" from the Spent Liquor 61 Isolation of the Cold Water Extract of Aspenwood 62 Determination of the Degree of Polymerization Hemicellulose 63 Alkali-Resistant Cellulose 64 Hydrolysis Procedures -

Chemical Resistance 100% SOLIDS EPOXY SYSTEMS
Chemical Resistance 100% SOLIDS EPOXY SYSTEMS CHEMICAL 8300 SYSTEM 8200 SYSTEM 8000 SYSTEM OVERKOTE PLUS HD OVERKOTE HD OVERKRETE HD BASED ON ONE YEAR IMMERSION TESTING –––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––– Acetic Acid (0-15%) G II Acetonitrile LLG L Continuous Immersion Acetone (0-20%) LLL Acetone (20-30%) Suitable for continuous immersion in that chemical (based on LLG Acetone (30-50%) L G I ONE YEAR testing) to assure unlimited service life. Acetone (50-100%) G II Acrylamide (0-50%) LLL G Short-Term Exposure Adipic Acid Solution LLL Alcohol, Isopropyl LLL Suitable for short-term exposure to that chemical such as Alcohol, Ethyl LLG secondary containment (72 hours) or splash and spill Alcohol, Methyl LLI (immediate clean-up). Allyl Chloride LLI Allylamine (0-20%) L L I Allylamine (20-30%) L G I I Not Suitable Allylamine (30-50%) GGI Not suitable for any exposure to that chemical. Aluminum Bromide LL– Aluminum Chloride L L – Aluminum Fluoride (0-25%) L L – This chart shows chemical resistance of our various Aluminum Hydroxide LLL 1 topping materials (90 mils – ⁄4"). These ratings are based on Aluminum Iodide LL– temperatures being ambient. At higher temperatures, chemical Aluminum Nitrate LL– resistance may be effected. When chemical exposure is Aluminum Sodium Chloride L L – minimal to non-existent, a 9000 System–FlorClad™ HD or Aluminum Sulfate LLL 4600 System– BriteCast™ HD may be used. Alums L L L 2-Aminoethoxyethanol Resistance data is listed with the assumption that the material GGG has properly cured for at least four days, at recommended Ammonia – Wet L L – temperatures, prior to any chemical exposure. -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -

Rowe Scientific Barium Chloride Dihydrate
ROWE SCIENTIFIC BARIUM CHLORIDE DIHYDRATE ROWE SCIENTIFIC Chemwatch Hazard Alert Code: 3 Chemwatch: 11379 Issue Date: 27/06/2017 Version No: 4.1.1.1 Print Date: 23/09/2019 Safety Data Sheet according to WHS and ADG requirements S.GHS.AUS.EN SECTION 1 IDENTIFICATION OF THE SUBSTANCE / MIXTURE AND OF THE COMPANY / UNDERTAKING Product Identifier Product name BARIUM CHLORIDE, DIHYDRATE Chemical Name barium chloride, dihydrate Synonyms CB0480, CB0481 Proper shipping name BARIUM COMPOUND, N.O.S. (contains barium chloride, dihydrate) Chemical formula BaCl2·2H2O|Ba-Cl2 Other means of Not Available identification CAS number 10326-27-9 Relevant identified uses of the substance or mixture and uses advised against Manufacture of pigments, colour lakes, glass, mordant for acid dyes; weighting and dyeing textile fabrics; in aluminium Relevant identified uses refining; as a pesticide; boiler compounds for softening water; tanning and finishing leather. Details of the supplier of the safety data sheet Registered company ROWE SCIENTIFIC name Address 11 Challenge Boulevard, Wangara WA 6065 Australia Telephone +61 8 9302 1911 Fax +61 8 9302 1905 Website Not Available Email [email protected] Emergency telephone number Association / ROWE SCIENTIFIC Organisation Emergency telephone +61 8 9302 1911 (24 Hrs) numbers Other emergency Not Available telephone numbers SECTION 2 HAZARDS IDENTIFICATION Classification of the substance or mixture HAZARDOUS CHEMICAL. DANGEROUS GOODS. According to the WHS Regulations and the ADG Code. Poisons Schedule S6 Classification [1] Acute Toxicity (Inhalation) Category 4, Acute Toxicity (Oral) Category 3 1. Classified by Chemwatch; 2. Classification drawn from HCIS; 3. Classification drawn from Regulation (EU) No 1272/2008 - Legend: Annex VI Label elements Continued.. -

Decomposition of Dithionates Dissertation
DECOMPOSITION OF DITHIONATE S DISSE RTATI ON PRESEN TE D IN PA RTIAL F%LFI L LM EN T O F TH E REQ %IREM ENTS FO R THE D EG REE O F DO C TO R O F PHI LO SO PH Y IN THE G RA D%A TE S C H O O L O F THE O HIO STATE %N I%ERSITY BY JACOB CORNOG 1921 TABLE O F C ONTE NTS R eview of th e Lit er atur e P ertin ent t o th e Form ation and f t t po sition o Di hiona es . Aim s of th e Pr es ent Work on of B ar i um Dithionate a x e e t al of V riou s Dithionates (E p rim n . ) G en eral C on sider ation s an d Proc edur e T he D ecompo s ition of Barium Dithion at e of Some Other Dithionat es th e Lit eratur e P LYT H I O A T E I . T H E O N S H Dithionic acid , 2 8 2 0 6 , the first ember of this remarkable 1 - group of acids , was discovered in 9 by Welter and Gay Lus 1 5“ sacl H 1 842 Lan lo i s z ; trithionic acid , 2 S 3 0 6 , in , by g ; tetrathionic H 1 843 G elis 3 a acid , 2 8 4 0 6 , in , by Fordos and ; pent thionic acid , H 8 0 1 845 Wacken roder“ hexathi 2 5 6 , in , by ; finally potassium K 1 88 5 onate , Q S 6 , was discovered in 8 by D ebu s as a part of ’ roder his classic investigation of Wacken s solution . -

Chemical Resistance Chart
Polyurethane Thermal-Chem Corp. High Performance Epoxy Topcoats Products Chemical Resistance Chart E56 E = Excellent G = Good F = Fair Clear surfacer Plus 3.0 DecoTop < = Occasional Spillage U = Do Not Use AcidGard VersiGard ArmorTred Resurfacer Resurfacer ArmorClad Resurfacer Re DecoThane ArmorBond SolventGard DecoTop P92 Chemical 750 755 757 728 731 736 705 748 1056 1060 Polyurethane Thermal-Chem Corp. High Performance Epoxy Topcoats Products Chemical Resistance Chart E = Excellent G = Good F = Fair Clear surfacer Plus 3.0 DecoTop < = Occasional Spillage U = Do Not Use AcidGard VersiGard ArmorTred Resurfacer Resurfacer ArmorClad Resurfacer Re ThermalCrete ArmorBond SolventGard DecoFinish P81 Chemical 750 755 757 728 731 736 705 748 1030 1062 1, 4-Dichloro-2-butene U E E U U U U U U U 1, 4-Dioxane U < < U U U U U U U 1-1-1 Trichloroethane E E E F F G G F G G < < < < < 2, 4-Pentanedione F E E F F 3, 4-Dichloro-1-butene < E E U U U U U U U 4-Picoline (0-50%) U < < U U U U U V V Acetic Acid (0-5%) E E E G G G G G E G < < < < < < Acetic Acid (5-15%) E E E F Acetone(0-20%) E E E F G G G G E F Acetone (20-30% < E E < < < < < E < Acetone (30-50%) < E E < < < < < E < Acetone (50-100%) < F F < < < < < E < Acetonitrile F E E < < < < < E < Acrylamide (0-50%) E E E F G G G G F Adipic Acid Solution E E E E E E E E G Alcohol, Ethyl E E E F F F F F F Alcohol, Isopropyl E E E F F F F F G F Alcohol, Methyl < E E < < < < < < < Allyl Chloride < E E U U U U U E < Allylamine (0-20%) < E E U U U U U < Allylamine (20-30%) < E E U U U U U < Allylamine (30-50%) U < < U U U U U U Aluminum Bromate E E E F G G G G E Aluminum Bromide E E E E E E E E E Aluminum Chloride E E E E E E E E E Aluminum Fluoride (25%) E E E E E E E E E Polyurethane Thermal-Chem Corp. -

HDPE Chemical Resistance Guide
HDPE Chemical Resistance Guide 70º F 140º F 70º F 140º F Reagent (21º C) (60º C) Reagent (21º C) (60º C) A B Acetaldehyde S O Barium carbonate saturated S S Acetic acid (1-10%) S S Barium carbonate saturated S S Acetic acid (10-60%) S O Barium hydroxide S S Acetic acid (80-100%) S O Barium sulfate saturated S S Acetic anhydride S S Barium sulfite saturated S S Acetone S S Beer S S Acids (aromatic) S S Benzaldehyde S O Acrylic emulsions S S Benzene O U Adipic acid S S Benzene sulfonic acid S S Aluminum chloride concentrated S S Benzoic acid crystals S S Aluminum chloride dilute S S Benzoic acid saturated S S Aluminum fluoride concentrated S S Bismuth carbonate saturated S S Aluminum sulfate concentrated S S Black liquor S S Alums (all types) concentrated S S Bleach lye (10%) S S Amino acetic acid S S Borax cold saturated S S Ammonia (100% dry gas) S S Boric acid concentrated S S Ammonium acetate S S Boric acid dilute S S Ammonium bromide S S Brine S S Ammonium carbonate S S Bromic acid (10%) S S Ammonium chloride saturated S S Bromine liquid (100%) O U Ammonium fluoride (20%) S S Bromochloromethane U U Ammonium hydroxide S S Butadiene U U Ammonium metaphosphate (sat.) S S Butanediol (10%) S S Ammonium nitrate saturated S S Butanediol (60%) S S Ammonium persulfate saturated S S Butanediol (100%) S S Ammonium phosphate S S Butter S S Ammonium sulfate saturated S S Butyl acetate (100%) O U Ammonium sulfide saturated S S Butyl alcohol (100%) S S Ammonium thiocyanate saturated S S Butylene glycol S S Amyl acetate (100%) O U Butyric acid (100%) -
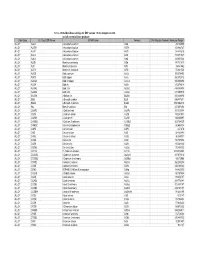
Database Full Listing
16-Nov-06 OLI Data Base Listings for ESP version 7.0.46, Analyzers 2.0.46 and all current alliance products Data Base OLI Tag (ESP) Name IUPAC Name Formula CAS Registry Number Molecular Weight ALLOY AL2U 2-Aluminum uranium Al2U 291.98999 ALLOY AL3TH 3-Aluminum thorium Al3Th 312.982727 ALLOY AL3TI 3-Aluminum titanium Al3Ti 128.824615 ALLOY AL3U 3-Aluminum uranium Al3U 318.971527 ALLOY AL4U 4-Aluminum uranium Al4U 345.953064 ALLOY ALSB Aluminum antimony AlSb 148.731537 ALLOY ALTI Aluminum titanium AlTi 74.861542 ALLOY ALTI3 Aluminum 3-titanium AlTi3 170.621536 ALLOY AUCD Gold cadmium AuCd 309.376495 ALLOY AUCU Gold copper AuCu 260.512512 ALLOY AUCU3 Gold 3-copper AuCu3 387.604492 ALLOY AUSN Gold tin AuSn 315.676514 ALLOY AUSN2 Gold 2-tin AuSn2 434.386505 ALLOY AUSN4 Gold 4-tin AuSn4 671.806519 ALLOY BA2SN 2-Barium tin Ba2Sn 393.369995 ALLOY BI2U 2-Bismuth uranium Bi2U 655.987671 ALLOY BI4U3 4-Bismuth 3-uranium Bi4U3 1550.002319 ALLOY BIU Bismuth uranium BiU 447.007294 ALLOY CA2PB 2-Calcium lead Ca2Pb 287.355988 ALLOY CA2SI 2-Calcium silicon Ca2Si 108.241501 ALLOY CA2SN 2-Calcium tin Ca2Sn 198.865997 ALLOY CA3SB2 3-Calcium 2-antimony Ca3Sb2 363.734009 ALLOY CAMG2 Calcium 2-magnesium CaMg2 88.688004 ALLOY CAPB Calcium lead CaPb 247.278 ALLOY CASI Calcium silicon CaSi 68.163498 ALLOY CASI2 Calcium 2-silicon CaSi2 96.249001 ALLOY CASN Calcium tin CaSn 158.787994 ALLOY CAZN Calcium zinc CaZn 105.468002 ALLOY CAZN2 Calcium 2-zinc CaZn2 170.858002 ALLOY CD11U 11-Cadmium uranium Cd11U 1474.536865 ALLOY CD3AS2 3-Cadmium 2-arsenic As2Cd3 487.073212 -

Barium Sulfite
240 Barium Sulfite COHPONE:-iTS: EVALUATOR: 1. Bar1um sulf1te; BaS03; [7787-39-5] H.D. Lutz, Dept. of Chem1stry, 2. Water; H20; [7732-18-5] University of Slegen, FR Germany. August 1983. CRITICAL EVALUATION: Bar1um sulflte crystalllzes from aqueous solutions 1n the form of the anhydrous salt, BaS03' The solub111ty of BaS03 1n water has not yet been thoroughly invest1gated. Numer1cal data have been reported by Autenrleth et a1. (1), ROgow1CZ (2), Parsons (3), Marecek et a1. (4), Rod1n ct a1. (5), and Cohen et a1. (6), yet not under directly comparable exper1mental conditions. In 1898, Autenr1eth et a1. (1) reported the value 4 of 0.022 g/kg H20 (m(BaS03) = 1.0 x 10- mol kg-I) at 289 K. Rogowicz (2) gave the values 0.1974 and 0.0177 g/dm3 (c(BaS03) = 9.080 x 10-4 and 8.14 x 10-5 mol dm- 3) at 293 and 353 K, respect1vely. Parsons (3) reported the solubil1ty product of BaS03 (K sO ) as 9.5 x 10-10 mol 2 dm-6 at 298.2 K from a source which is not given nor could be traced. This value 1S equ1valent to a solubility of 6.7 mg/dm3 (c(BaS03) = 3.08 x 10-5 mol dm- 3). In 1983 Rod1n et a1. (5) determined some values from 1.23 x 10-3 g/dm3 (c(BaS03) = 5.7 x 10-6 mol dm- 3) at 293 K to 3.5 x 10-3 g/dm3 (1.61 x 10-5 mol dm-3) at 353 K. -

Solubility Product Constants
SOLUBILITY PRODUCT CONSTANTS The solubility product constant Ksp is a useful parameter for which is strongly shifted to the right except in very basic solutions. calculating the aqueous solubility of sparingly soluble compounds Furthermore, the equilibrium constant for this reaction, which de- under various conditions. It may be determined by direct measure- pends on the second ionization constant of H2S, is poorly known. ment or calculated from the standard Gibbs energies of formation Therefore it is more useful in the case of sulfides to define a differ- ∆fG° of the species involved at their standard states. Thus if Ksp = ent solubility product Kspa based on the reaction [M+]m, [A–]n is the equilibrium constant for the reaction + + MmSn(s) + 2H mM + nH2S (aq) + – MmAn(s) mM (aq) + nA (aq), Values of Kspa , taken from Reference 8, are given for several sulfides + - where MmAn is the slightly soluble substance and M and A are the in the auxiliary table following the main table. Additional discus- ions produced in solution by the dissociation of MmAn, then the sion of sulfide equilibria may be found in References 7 and 9. Gibbs energy change is + – ∆G° = m ∆fG° (M ,aq) + n ∆fG° (A ,aq) –∆fG° (MmAn, s) References The solubility product constant is calculated from the equation 1. Wagman, D. D., Evans, W. H., Parker, V. B., Schumm, R. H., Halow, I., Bailey, S. M., Churney, K. L., and Nuttall, R. L., The NBS Tables of Chemical Thermodynamic Properties, J. Phys. Chem. Ref. Data, Vol. ln K = –∆ G°/RT sp 11, Suppl. 2, 1982. -

Lawrence Berkeley National Laboratory Recent Work
Lawrence Berkeley National Laboratory Recent Work Title GEOCHEMICAL AND COSMOCHEMICAL CYCLES INVOLVING SULFUR, SULFIDE, SULFITE, AND SULFATE Permalink https://escholarship.org/uc/item/3jb1c1qd Author Meyer, B. Publication Date 1979-07-01 eScholarship.org Powered by the California Digital Library University of California LBL-9396 (!_~ Preprint · lrnl Lawrence Berkeley Laboratory ~ UNIVERSITY OF CALIFORNIA, BERKELEY, CA Materials & Molecular Research Division Submitted to Geochimica Cosmochimica Acta GEOCHEMICAL AND COSMOCHEMICAL CYCLES INVOLVING SULFUR, SULFIDE, SULFITE, AND SULFATE -. ECEP/ED B. Meyer, L. Peter and M. Ospina ~o./l.WR!:NCE Sl:fi..KW,'ZY I.A30RAiORY ! IJI 7 Q HY"Ig July 1979 .J t L u 1::1/ LlBR!.RY AND DOCUMENTS SE:Cl'IO.N TWp-WEEK LOAN COPY This is a Library Circulating Copy which may be borrowed for two weeks. For a personal retention copy, call Tech. Info. Division, Ext. 6782 Prepared for the U. S. Department of Energy under Contract W-7405-ENG-48 DISCLAIMER This document was prepared as an account of work sponsored by the United States Government. While this document is believed to contain correct information, neither the United States Government nor any agency thereof, nor the Regents of the University of California, nor any of their employees, makes any warranty, express or implied, or assumes any legal responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by its trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof, or the Regents of the University of California.