Photochemical Reaction Products of Polycyclic Aromatic Hydrocarbons
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Vibrationally Excited Hydrogen Halides : a Bibliography On
VI NBS SPECIAL PUBLICATION 392 J U.S. DEPARTMENT OF COMMERCE / National Bureau of Standards National Bureau of Standards Bldg. Library, _ E-01 Admin. OCT 1 1981 191023 / oO Vibrationally Excited Hydrogen Halides: A Bibliography on Chemical Kinetics of Chemiexcitation and Energy Transfer Processes (1958 through 1973) QC 100 • 1X57 no. 2te c l !14 c '- — | NATIONAL BUREAU OF STANDARDS The National Bureau of Standards' was established by an act of Congress March 3, 1901. The Bureau's overall goal is to strengthen and advance the Nation's science and technology and facilitate their effective application for public benefit. To this end, the Bureau conducts research and provides: (1) a basis for the Nation's physical measurement system, (2) scientific and technological services for industry and government, (3) a technical basis for equity in trade, and (4) technical services to promote public safety. The Bureau consists of the Institute for Basic Standards, the Institute for Materials Research, the Institute for Applied Technology, the Institute for Computer Sciences and Technology, and the Office for Information Programs. THE INSTITUTE FOR BASIC STANDARDS provides the central basis within the United States of a complete and consistent system of physical measurement; coordinates that system with measurement systems of other nations; and furnishes essential services leading to accurate and uniform physical measurements throughout the Nation's scientific community, industry, and commerce. The Institute consists of a Center for Radiation Research, an Office of Meas- urement Services and the following divisions: Applied Mathematics — Electricity — Mechanics — Heat — Optical Physics — Nuclear Sciences" — Applied Radiation 2 — Quantum Electronics 1 — Electromagnetics 3 — Time 3 1 1 and Frequency — Laboratory Astrophysics — Cryogenics . -

Organic Data Validation Report Soil Samples Collected June 4,1997 Brodhead Creek, Stroudsburg, Pa
ENVIRONMENTAL RESEARCH AND CONSULTING, INC. 112 COMMONS COURT CHADDS FORD, PA 19317 ORGANIC DATA VALIDATION REPORT SOIL SAMPLES COLLECTED JUNE 4,1997 BRODHEAD CREEK, STROUDSBURG, PA JULY 1997 ACRES INTERNATIONAL CORPORATION 140 John James Audubon Parkway Amherst, New York 14228-1 1 80 AR302871* TABLE OF CONTENTS 1 NARRATIVE ........................................................... 1 2 OVERVIEW ............................................................ 1 3 SUMMARY ........................................................... 1 4 MAJORPROBLEMS .................................................... 1 5 MINOR PROBLEMS .................................................... 2 8 NOTES ......................................................."........ 2 7 REPORT CONTENT .................................................... 2 8 ATTACHMENTS ......".......'.......................................... 3 APPENDICES APPENDIX A - GLOSSARY OF DATA QUALIFIER CODES APPENDIX B - DATA SUMMARY FORMS^ APPENDIX C- RESULTS AS REPORTED BY THE LABORATORY FOR ALL TARGET COMPOUNDS APPENDIX D - REVIEWED AND ACCEPTED TENTATIVELY IDENTIFIED COMPOUNDS APPENDIX E - SUPPORT DOCUMENTATION AR302875 1 NARRATIVE Date: July 28,1997 Subject Organic Data Validation for Sample Delivery Group (SDG) #BRH01 Brodhead Creek • Stroudsburg, Pennsylvania From: James R. Stachowski. Environmental Specialist Acres International Corporation To: Harold M. Brundage III Environmental Research and Consulting, Inc. 2 OVERVIEW This report pertains to data validation of fifteen (15) soil samples from the -

ORGANIC CHEMISTRY- I (Nature of Title Bonding and Stereochemistry) Module No
Subject Chemistry Paper No. and Paper 1: ORGANIC CHEMISTRY- I (Nature of Title Bonding and Stereochemistry) Module No. and Module 8: Aromaticity of fused rings Title Module Tag CHE_P1_M8 CHEMISTRY PAPER 1: ORGANIC CHEMISTRY- I(Nature of Bonding and Stereochemistry) MODULE 8: Aromaticity of fused rings TABLE OF CONTENT 1. Learning Outcomes 2. Introduction 3. Classification of fused ring systems 4. Aromaticity in fused ring systems 4.1. Aromaticity of some benzenoid fused systems 4.1.1. Naphthalene 4.1.2. Anthracene 4.1.3. Phenanthrene 4.1.4. Resonance energy of fused ring systems 4.2. Aromaticity of some non-benzenoid fused systems 4.2.1. Azulenes 4.2.2. Oxaazulenaones 5. Other fused ring systems 5.1. Phenalene 5.2. Benzo cyclobutadiene 5.3. Ferrocene 6. Summary CHEMISTRY PAPER 1: ORGANIC CHEMISTRY- I(Nature of Bonding and Stereochemistry) MODULE 8: Aromaticity of fused rings 1. Learning Outcomes After studying this module, you shall be able to: Learn about the fused rings Understand that how fused rings are classified Learn about the aromaticity of the fused rings Understand aromaticity in the benzenoid and non-benzenoid fused ring systems Learn about some other special cases 2. Introduction As you are already aware that the aromatic compounds apparently contain alternate double and single bonds in a cyclic structure and resemble benzene in chemical behavior. Up till now we have discussed the aromaticity in monocyclic rings. In this module, we shall discuss about the aromaticity of fused rings. So, before starting with the aromaticity of fused rings first we should know what fused rings are. -

UNITED STATES PATENT of FICE 2,640,086 PROCESS for SEPARATING HYDROGEN FLUORIDE from CHLORODFLUORO METHANE Robert H
Patented May 26, 1953 2,640,086 UNITED STATES PATENT of FICE 2,640,086 PROCESS FOR SEPARATING HYDROGEN FLUORIDE FROM CHLORODFLUORO METHANE Robert H. Baldwin, Chadds Ford, Pa., assignor to E. H. du Pont de Nemours and Company, Wi inington, Del, a corporation of Delaware No Drawing. Application December 15, 1951, Serial No. 261,929 9 Claims. (C. 260-653) 2 This invention relates to a process for Sep These objects are accomplished essentially by arating hydrogen fluoride from monochlorodi Subjecting a mixture of hydrogen fluoride and fluoronethane, and more particularly, separat Inonochlorodifluoromethane in the liquid phase ing these components from the reaction mixture to temperatures below 0° C., preferably at about obtained in the fluorination of chloroform with -30° C. to -50° C., at either atmospheric or hydrogen fluoride, Super-atmospheric pressures, together with from In the fluorination of chloroform in the prest about 0.25 mol to about 2.5 mols of chloroform ence Of a Catalyst, a reaction mixture is pro per mol of chlorodifluoronethane contained in duced which consists essentially of HCl, HF, the mixture and separating an upper layer rich CHCIF2, CHCl2F, CHCls, and CHF3. A method O in HF from a lower organic layer. The proceSS of Separating these components is disclosed in is operative with mixtures containing up to 77% U. S. Patent No. 2,450,414 which involves sep by weight of HF. arating the components by a special fractional It has been found that chloroform is substan distillation under appropriate temperatures and tially immiscible With EIF at temperatures be pressures. -
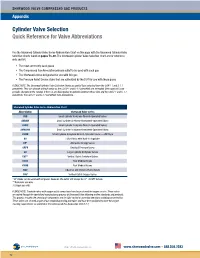
Cylinder Valve Selection Quick Reference for Valve Abbreviations
SHERWOOD VALVE COMPRESSED GAS PRODUCTS Appendix Cylinder Valve Selection Quick Reference for Valve Abbreviations Use the Sherwood Cylinder Valve Series Abbreviation Chart on this page with the Sherwood Cylinder Valve Selection Charts found on pages 73–80. The Sherwood Cylinder Valve Selection Chart are for reference only and list: • The most commonly used gases • The Compressed Gas Association primary outlet to be used with each gas • The Sherwood valves designated for use with this gas • The Pressure Relief Device styles that are authorized by the DOT for use with these gases PLEASE NOTE: The Sherwood Cylinder Valve Selection Charts are partial lists extracted from the CGA V-1 and S-1.1 pamphlets. They can change without notice as the CGA V-1 and S-1.1 pamphlets are amended. Sherwood will issue periodic changes to the catalog. If there is any discrepancy or question between these lists and the CGA V-1 and S-1.1 pamphlets, the CGA V-1 and S-1.1 pamphlets take precedence. Sherwood Cylinder Valve Series Abbreviation Chart Abbreviation Sherwood Valve Series AVB Small Cylinder Acetylene Wrench-Operated Valves AVBHW Small Cylinder Acetylene Handwheel-Operated Valves AVMC Small Cylinder Acetylene Wrench-Operated Valves AVMCHW Small Cylinder Acetylene Handwheel-Operated Valves AVWB Small Cylinder Acetylene Wrench-Operated Valves — WB Style BV Hi/Lo Valves with Built-in Regulator DF* Alternative Energy Valves GRPV Residual Pressure Valves GV Large Cylinder Acetylene Valves GVT** Vertical Outlet Acetylene Valves KVAB Post Medical Valves KVMB Post Medical Valves NGV Industrial and Chrome-Plated Valves YVB† Vertical Outlet Oxygen Valves 1 * DF Valves can be used with all gases; however, the outlet will always be ⁄4"–18 NPT female. -

Ammonium Bifluoride CAS No
Product Safety Summary Ammonium Bifluoride CAS No. 1341-49-7 This Product Safety Summary is intended to provide a general overview of the chemical substance. The information on the summary is basic information and is not intended to provide emergency response information, medical information or treatment information. The summary should not be used to provide in-depth safety and health information. In-depth safety and health information can be found in the Safety Data Sheet (SDS) for the chemical substance. Names • Ammonium bifluoride (ABF) • Ammonium difluoride • Ammonium acid fluoride • Ammonium hydrogen difluoride • Ammonium fluoride compound with hydrogen fluoride (1:1) Product Overview Solvay Fluorides, LLC does not sell ammonium bifluoride directly to consumers. Ammonium bifluoride is used in industrial applications and in other processes where workplace exposures can occur. Ammonium bifluoride (ABF) is used for cleaning and etching of metals before they are further processed. It is used as an oil well acidifier and in the etching of glass or cleaning of brick and ceramics. It may also be used for pH adjustment in industrial textile processing or laundries. ABF is available as a solid or liquid solution (in water). Ammonium bifluoride is a corrosive chemical and contact can severely irritate and burn the skin and eyes causing possible permanent eye damage. Breathing ammonium bifluoride can severely irritate and burn the nose, throat, and lungs, causing nosebleeds, cough, wheezing and shortness of breath. On contact with water or moist skin, ABF can release hydrofluoric acid, a very dangerous acid. Inhalation or ingestion of large amounts of ammonium bifluoride can cause nausea, vomiting and loss of appetite. -

Reactivity Landscape of Pyruvate Under Simulated Hydrothermal Vent
Reactivity landscape of pyruvate under simulated SEE COMMENTARY hydrothermal vent conditions Yehor Novikova and Shelley D. Copleyb,c,1 aDepartment of Chemistry and Biochemistry, bDepartment of Molecular, Cellular, and Developmental Biology, and cCooperative Institute for Research in Environmental Sciences, University of Colorado Boulder, Boulder, CO 80309 Edited by Paul G. Falkowski, Rutgers, The State University of New Jersey, New Brunswick, NJ, and approved June 14, 2013 (received for review March 14, 2013) Pyruvate is an important “hub” metabolite that is a precursor for concentrations of many components (4). Fig. 1 shows an example in amino acids, sugars, cofactors, and lipids in extant metabolic net- which the availability of catalysts for different steps in a network works. Pyruvate has been produced under simulated hydrother- results in significantly different network topologies and accumu- mal vent conditions from alkyl thiols and carbon monoxide in the lation of different products. Network topology also depends on the presence of transition metal sulfides at 250 °C [Cody GD et al. set of reagents available and the concentrations of those reagents. K (2000) Science 289(5483):1337–1340], so it is plausible that pyru- For example, the network depicted in Fig. 1 would form only and M H J M vate was formed in hydrothermal systems on the early earth. We if no were available, and would form only and if the concentration of H were very high (assuming equal rate constants report here that pyruvate reacts readily in the presence of transi- D tion metal sulfide minerals under simulated hydrothermal vent for the partitioning of between the two possible pathways). -

Introduction to Ionic Mechanisms Part I: Fundamentals of Bronsted-Lowry Acid-Base Chemistry
INTRODUCTION TO IONIC MECHANISMS PART I: FUNDAMENTALS OF BRONSTED-LOWRY ACID-BASE CHEMISTRY HYDROGEN ATOMS AND PROTONS IN ORGANIC MOLECULES - A hydrogen atom that has lost its only electron is sometimes referred to as a proton. That is because once the electron is lost, all that remains is the nucleus, which in the case of hydrogen consists of only one proton. The large majority of organic reactions, or transformations, involve breaking old bonds and forming new ones. If a covalent bond is broken heterolytically, the products are ions. In the following example, the bond between carbon and oxygen in the t-butyl alcohol molecule breaks to yield a carbocation and hydroxide ion. H3C CH3 H3C OH H3C + OH CH3 H3C A tertiary Hydroxide carbocation ion The full-headed curved arrow is being used to indicate the movement of an electron pair. In this case, the two electrons that make up the carbon-oxygen bond move towards the oxygen. The bond breaks, leaving the carbon with a positive charge, and the oxygen with a negative charge. In the absence of other factors, it is the difference in electronegativity between the two atoms that drives the direction of electron movement. When pushing arrows, remember that electrons move towards electronegative atoms, or towards areas of electron deficiency (positive, or partial positive charges). The electron pair moves towards the oxygen because it is the more electronegative of the two atoms. If we examine the outcome of heterolytic bond cleavage between oxygen and hydrogen, we see that, once again, oxygen takes the two electrons because it is the more electronegative atom. -

Hexafluorosilicic Acid
Sodium Hexafluorosilicate [CASRN 16893-85-9] and Fluorosilicic Acid [CASRN 16961-83-4] Review of Toxicological Literature October 2001 Sodium Hexafluorosilicate [CASRN 16893-85-9] and Fluorosilicic Acid [CASRN 16961-83-4] Review of Toxicological Literature Prepared for Scott Masten, Ph.D. National Institute of Environmental Health Sciences P.O. Box 12233 Research Triangle Park, North Carolina 27709 Contract No. N01-ES-65402 Submitted by Karen E. Haneke, M.S. (Principal Investigator) Bonnie L. Carson, M.S. (Co-Principal Investigator) Integrated Laboratory Systems P.O. Box 13501 Research Triangle Park, North Carolina 27709 October 2001 Toxicological Summary for Sodium Hexafluorosilicate [16893-85-9] and Fluorosilicic Acid [16961-83-4] 10/01 Executive Summary Nomination Sodium hexafluorosilicate and fluorosilicic acid were nominated for toxicological testing based on their widespread use in water fluoridation and concerns that if they are not completely dissociated to silica and fluoride in water that persons drinking fluoridated water may be exposed to compounds that have not been thoroughly tested for toxicity. Nontoxicological Data Analysis and Physical-Chemical Properties Analytical methods for sodium hexafluorosilicate include the lead chlorofluoride method (for total fluorine) and an ion-specific electrode procedure. The percentage of fluorosilicic acid content for water supply service application can be determined by the specific-gravity method and the hydrogen titration method. The American Water Works Association (AWWA) has specified that fluorosilicic acid contain 20 to 30% active ingredient, a maximum of 1% hydrofluoric acid, a maximum of 200 mg/kg heavy metals (as lead), and no amounts of soluble mineral or organic substance capable of causing health effects. -

Coulomb Pairing Resonances in Multiple-Ring Aromatic Molecules
Coulomb pairing resonances in multiple-ring aromatic molecules D.L. Huber* Physics Department, University of Wisconsin-Madison, Madison, Wisconsin 53706, USA Abstract We present an analysis of pairing resonances observed in photo-double-ionization studies of CnHm aromatic molecules with multiple benzene-like rings. The analysis, which is based on the Coulomb pairing model, is applied to naphthalene, anthracene, phenanthrene, pyrene and coronene, all of which have six-member rings, and azulene which is comprised of a five-member and a seven-member ring. There is a high energy resonance at ~ 40 eV that is found in all of the molecules cited and is associated with paired electrons localized on carbon sites on the perimeter of the molecule, each of which having two carbon sites as nearest neighbors. The low energy resonance at 10 eV, which is found only in pyrene and coronene, is attributed to the formation of paired HOMO electrons localized on arrays of interior carbon atoms that have the point symmetry of the molecule with each carbon atom having three nearest neighbors. The origin of the anomalous increase in the doubly charged to singly charged parent-ion ratio that is found above the 40 eV resonance in all of the cited molecules except coronene is discussed. *Mailing address: Physics Department, University of Wisconsin-Madison, 1150 University Ave., Madison, WI 53711, USA; e-mail: [email protected] 1 1. Introduction Recent studies of photo-double-ionization in CnHm multiple-ring (polycylic) aromatic molecules have revealed the existence of anomalous resonances in the ratio of the cross sections of doubly charged parent ions to singly charged parent ions I(2+)/I(1+) [1-4]. -

Occupational Exposure to Hydrogen Fluoride
criteria for a recommended standard OCCUPATIONAL EXPOSURE TO HYDROGEN FLUORIDE U.S. DEPARTMENT OF HEALTH, EDUCATION, AND WELFARE Public Health Service Center for Disease Control National Institute for Occupational Safety and Health M arch 1976 HEW Publication No. (NIOSH) 7 6 -1 4 3 PREFACE The Occupational Safety and Health Act of 1970 emphasizes the need for standards to protect the health and safety of workers exposed to an ever-increasing number of potential hazards at their workplace. The National Institute for Occupational Safety and Health has projected a formal system of research, with priorities determined on the basis of specified indices, to provide relevant data from which valid criteria for effective standards can be derived. Recommended standards for occupational exposure, which are the result of this work, are based on the health effects of exposure. The Secretary of Labor will weigh these recommen dations along with other considerations such as feasibility and means of implementation in developing regulatory standards. It is intended to present successive reports as research and epide miologic studies are completed and as sampling and analytical methods are developed. Criteria and standards will be reviewed periodically to ensure continuing protection of the worker. I am pleased to acknowledge the contributions to this report on hydrogen fluoride by members of my staff and the valuable constructive comments by the Review Consultants on Hydrogen Fluoride, by the ad hoc committees of the American Academy of Occupational Medicine and the Society for Occupational and Environmental Health, and by Robert B. O'Connor, M.D., NIOSH consultant in occupational medicine. -

Anti-Coking Materials for Steam Crackers Copyright
ANTI-COKING MATERIALS FOR STEAM CRACKERS A Dissertation Presented to The Academic Faculty by Shilpa Mahamulkar In Partial Fulfillment of the Requirements for the Degree DOCTOR of PHILOSOPHY in the SCHOOL OF CHEMICAL AND BIOMOLECULAR ENGINEERING Georgia Institute of Technology May 2017 COPYRIGHT © 2017 BY SHILPA MAHAMULKAR ANTI COKING MATERIALS FOR STEAM CRACKERS Approved by: Prof. Christopher W. Jones, Advisor Prof. Athanasios Nenes School of Chemical & Biomolecular School of Earth & Atmospheric Engineering Sciences Georgia Institute of Technology Georgia Institute of Technology Prof. Pradeep K. Agrawal, Co-advisor Dr. Andrzej Malek School of Chemical & Biomolecular Hydrocarbons R&D, Midland Engineering The Dow Chemical Company Georgia Institute of Technology Prof. Thomas Fuller School of Chemical & Biomolecular Engineering Georgia Institute of Technology Date Approved: 20th March, 2017 To my wonderful parents Suresh and Vasanti Mahamulkar & my loving husband Ravi Kumar Kovvali ACKNOWLEDGEMENTS Foremost, I would like to extend my sincere thanks to my advisors Dr. Christopher Jones and Dr. Pradeep Agrawal for their continuous support throughout these five years. I am grateful for their invaluable advice, constructive criticism and the positive appreciation. Their enthusiasm and dedication towards work has been really inspiring. I thank the Dow Chemical Company, for funding the project and giving me an opportunity to acquire hands on experience in an industrial setting. I would like to thank our collaborators from University of Virginia, Prof. Robert Davis and Dr. Kehua Yin for the fruitful discussions and suggestions which have been instrumental in the work. I had the pleasure to work with Dr. Hyuk Taek Kwon and would like to thank him for mentoring me in a new field of coatings.