First-Principles Phase Stability, Bonding, and Electronic Structure of Actinide Metals
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Actinide Ground-State Properties-Theoretical Predictions
Actinide Ground-State Properties Theoretical predictions John M. Wills and Olle Eriksson electron-electron correlations—the electronic energy of the ground state of or nearly fifty years, the actinides interactions among the 5f electrons and solids, molecules, and atoms as a func- defied the efforts of solid-state between them and other electrons—are tional of electron density. The DFT Ftheorists to understand their expected to affect the bonding. prescription has had such a profound properties. These metals are among Low-symmetry crystal structures, impact on basic research in both the most complex of the long-lived relativistic effects, and electron- chemistry and solid-state physics that elements, and in the solid state, they electron correlations are very difficult Walter Kohn, its main inventor, was display some of the most unusual to treat in traditional electronic- one of the recipients of the 1998 behaviors of any series in the periodic structure calculations of metals and, Nobel Prize in Chemistry. table. Very low melting temperatures, until the last decade, were outside the In general, it is not possible to apply large anisotropic thermal-expansion realm of computational ability. And DFT without some approximation. coefficients, very low symmetry crystal yet, it is essential to treat these effects But many man-years of intense research structures, many solid-to-solid phase properly in order to understand the have yielded reliable approximate transitions—the list is daunting. Where physics of the actinides. Electron- expressions for the total energy in does one begin to put together an electron correlations are important in which all terms, except for a single- understanding of these elements? determining the degree to which 5f particle kinetic-energy term, can be In the last 10 years, together with electrons are localized at lattice sites. -
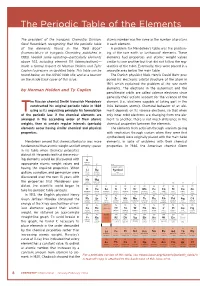
The Periodic Table of the Elements
The Periodic Table of the Elements The president of the Inorganic Chemistry Division, atomic number was the same as the number of protons Gerd Rosenblatt, recognizing that the periodic table in each element. of the elements found in the “Red Book” A problem for Mendeleev’s table was the position- (Nomenclature of Inorganic Chemistry, published in ing of the rare earth or lanthanoid* elements. These 1985) needed some updating—particularly elements elements had properties and atomic weight values above 103, including element 110 (darmstadtium)— similar to one another but that did not follow the reg- made a formal request to Norman Holden and Tyler ularities of the table. Eventually, they were placed in a Coplen to prepare an updated table. This table can be separate area below the main table. found below, on the IUPAC Web site, and as a tear-off The Danish physicist Niels Henrik David Bohr pro- on the inside back cover of this issue. posed his electronic orbital structure of the atom in 1921, which explained the problem of the rare earth by Norman Holden and Ty Coplen elements. The electrons in the outermost and the penultimate orbits are called valence electrons since generally their actions account for the valence of the he Russian chemist Dmitri Ivanovich Mendeleev element (i.e., electrons capable of taking part in the constructed his original periodic table in 1869 links between atoms). Chemical behavior of an ele- Tusing as its organizing principle his formulation ment depends on its valence electrons, so that when of the periodic law: if the chemical elements are only inner orbit electrons are changing from one ele- arranged in the ascending order of their atomic ment to another, there is not much difference in the weights, then at certain regular intervals (periods) chemical properties between the elements. -

ELECTRONEGATIVITY D Qkq F=
ELECTRONEGATIVITY The electronegativity of an atom is the attracting power that the nucleus has for it’s own outer electrons and those of it’s neighbours i.e. how badly it wants electrons. An atom’s electronegativity is determined by Coulomb’s Law, which states, “ the size of the force is proportional to the size of the charges and inversely proportional to the square of the distance between them”. In symbols it is represented as: kq q F = 1 2 d 2 where: F = force (N) k = constant (dependent on the medium through which the force is acting) e.g. air q1 = charge on an electron (C) q2 = core charge (C) = no. of protons an outer electron sees = no. of protons – no. of inner shell electrons = main Group Number d = distance of electron from the nucleus (m) Examples of how to calculate the core charge: Sodium – Atomic number 11 Electron configuration 2.8.1 Core charge = 11 (no. of p+s) – 10 (no. of inner e-s) +1 (Group i) Chlorine – Atomic number 17 Electron configuration 2.8.7 Core charge = 17 (no. of p+s) – 10 (no. of inner e-s) +7 (Group vii) J:\Sciclunm\Resources\Year 11 Chemistry\Semester1\Notes\Atoms & The Periodic Table\Electronegativity.doc Neon holds onto its own electrons with a core charge of +8, but it can’t hold any more electrons in that shell. If it was to form bonds, the electron must go into the next shell where the core charge is zero. Group viii elements do not form any compounds under normal conditions and are therefore given no electronegativity values. -

Periodic Table of the Elements Notes
Periodic Table of the Elements Notes Arrangement of the known elements based on atomic number and chemical and physical properties. Divided into three basic categories: Metals (left side of the table) Nonmetals (right side of the table) Metalloids (touching the zig zag line) Basic Organization by: Atomic structure Atomic number Chemical and Physical Properties Uses of the Periodic Table Useful in predicting: chemical behavior of the elements trends properties of the elements Atomic Structure Review: Atoms are made of protons, electrons, and neutrons. Elements are atoms of only one type. Elements are identified by the atomic number (# of protons in nucleus). Energy Levels Review: Electrons are arranged in a region around the nucleus called an electron cloud. Energy levels are located within the cloud. At least 1 energy level and as many as 7 energy levels exist in atoms Energy Levels & Valence Electrons Energy levels hold a specific amount of electrons: 1st level = up to 2 2nd level = up to 8 3rd level = up to 8 (first 18 elements only) The electrons in the outermost level are called valence electrons. Determine reactivity - how elements will react with others to form compounds Outermost level does not usually fill completely with electrons Using the Table to Identify Valence Electrons Elements are grouped into vertical columns because they have similar properties. These are called groups or families. Groups are numbered 1-18. Group numbers can help you determine the number of valence electrons: Group 1 has 1 valence electron. Group 2 has 2 valence electrons. Groups 3–12 are transition metals and have 1 or 2 valence electrons. -

The Elements.Pdf
A Periodic Table of the Elements at Los Alamos National Laboratory Los Alamos National Laboratory's Chemistry Division Presents Periodic Table of the Elements A Resource for Elementary, Middle School, and High School Students Click an element for more information: Group** Period 1 18 IA VIIIA 1A 8A 1 2 13 14 15 16 17 2 1 H IIA IIIA IVA VA VIAVIIA He 1.008 2A 3A 4A 5A 6A 7A 4.003 3 4 5 6 7 8 9 10 2 Li Be B C N O F Ne 6.941 9.012 10.81 12.01 14.01 16.00 19.00 20.18 11 12 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 3 Na Mg IIIB IVB VB VIB VIIB ------- VIII IB IIB Al Si P S Cl Ar 22.99 24.31 3B 4B 5B 6B 7B ------- 1B 2B 26.98 28.09 30.97 32.07 35.45 39.95 ------- 8 ------- 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 4 K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr 39.10 40.08 44.96 47.88 50.94 52.00 54.94 55.85 58.47 58.69 63.55 65.39 69.72 72.59 74.92 78.96 79.90 83.80 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 5 Rb Sr Y Zr NbMo Tc Ru Rh PdAgCd In Sn Sb Te I Xe 85.47 87.62 88.91 91.22 92.91 95.94 (98) 101.1 102.9 106.4 107.9 112.4 114.8 118.7 121.8 127.6 126.9 131.3 55 56 57 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 6 Cs Ba La* Hf Ta W Re Os Ir Pt AuHg Tl Pb Bi Po At Rn 132.9 137.3 138.9 178.5 180.9 183.9 186.2 190.2 190.2 195.1 197.0 200.5 204.4 207.2 209.0 (210) (210) (222) 87 88 89 104 105 106 107 108 109 110 111 112 114 116 118 7 Fr Ra Ac~RfDb Sg Bh Hs Mt --- --- --- --- --- --- (223) (226) (227) (257) (260) (263) (262) (265) (266) () () () () () () http://pearl1.lanl.gov/periodic/ (1 of 3) [5/17/2001 4:06:20 PM] A Periodic Table of the Elements at Los Alamos National Laboratory 58 59 60 61 62 63 64 65 66 67 68 69 70 71 Lanthanide Series* Ce Pr NdPmSm Eu Gd TbDyHo Er TmYbLu 140.1 140.9 144.2 (147) 150.4 152.0 157.3 158.9 162.5 164.9 167.3 168.9 173.0 175.0 90 91 92 93 94 95 96 97 98 99 100 101 102 103 Actinide Series~ Th Pa U Np Pu AmCmBk Cf Es FmMdNo Lr 232.0 (231) (238) (237) (242) (243) (247) (247) (249) (254) (253) (256) (254) (257) ** Groups are noted by 3 notation conventions. -

Reactions of Lanthanides and Actinides in Molten Alkali Metal/Polychalcogenide Fluxes at Intermediate Temperatures (250-600 °C) Anthony C
Chem. Mater. 1997, 9, 387-398 387 Reactions of Lanthanides and Actinides in Molten Alkali Metal/Polychalcogenide Fluxes at Intermediate Temperatures (250-600 °C) Anthony C. Sutorik and Mercouri G. Kanatzidis* Department of Chemistry and Center for Fundamental Materials Research, Michigan State University, East Lansing, Michigan 48824 Received August 26, 1996X From the reaction of the elemental lanthanides and actinides in molten alkali metal/ polychalcogenide salts, several new ternary compounds have been discovered. Specifically, these phases are K4USe8 (I) and ALnQ4 (II), where A ) K, Ln ) Ce or Tb, and Q ) Se or A ) Rb, Ln ) Ce, Q ) Te; and NaLnS3 (III), where Ln ) La, Ce. The K4USe8 crystallizes in the orthorhombic space group Fdd2 (No. 43) with a ) 17.331(4) Å, b ) 20.584(3) Å, c ) 8.811(3) Å, Z ) 8. The KCeSe4 crystallizes in the tetragonal space group P4/nbm (No. 125) with a ) 6.376(2) Å, c ) 8.327(1) Å, Z ) 2. The KTbSe4 crystallizes in the tetragonal space group P4/nbm (No. 125) with a ) 6.255(2) Å, c ) 8.227(1) Å, Z ) 2. The RbCeTe4 crystallizes in the tetragonal space group P4/nbm (No. 125) with a ) 6.952(3) Å, c ) 9.084(4) Å, Z ) 2. The NaCeS3 crystallizes in the orthorhombic space group Pmmn (No. 59) with a ) 5.683(1) Å, b ) 4.238(2) Å, c ) 9.802(2) Å, Z ) 2. The NaLaS3 crystallizes in the orthorhombic space group Pmmn (No. 59) with a ) 5.752(4) Å, b ) 4.2796(6) Å, c ) 9.841(2) Å, Z ) 2. -

Actinide Separation Chemistry in Nuclear Waste Streams and Materials, December 1997
NEA/NSC/DOC(97)19 NEA NUCLEAR SCIENCE COMMITTEE ACTINIDE SEPARATION CHEMISTRY IN NUCLEAR WASTE STREAMS AND MATERIALS December 1997 NUCLEAR ENERGY AGENCY ORGANISATION FOR ECONOMIC CO-OPERATION AND DEVELOPMENT FOREWORD The separation of actinide elements from various waste materials is a significant problem facing developed countries. The issue arises primarily because of the potential long-term hazard of many of the actinides, but is also due to the regulatory requirements associated with actinide waste disposal, which are different from those associated with other radioactive wastes. The different regulations are in turn related to the different health hazards and generally longer half lives of the actinides. This issue is of continuing interest to the OECD/NEA Member countries primarily in relation to waste produced within power station fuel recycle. Similar problems exist for waste produced in the past as a result of nuclear weapons production programmes and wastes likely to be produced in the future emerging from operations required for waste disposal. The Nuclear Science Committee, representing the interests of the basic R&D in the OECD/NEA Member countries, has established a task force of experts in actinide chemistry to review the current and developing separation techniques and chemical processes of interest in separating actinides. This task force was commissioned with the preparation of a report on the subject. The task force members are listed in Annex 1. This report is intended to provide a timely and representative guide to technical journals and other sources of separation chemistry information on the actinide elements. An important objective is to provide information that may be used to conserve energy and protect humans and the environment. -

The Chemical Interactions of Actinides in the Environment
The Chemical Interactions of Actinides in the Environment Wolfgang Runde rom a chemist’s perspective, the more oxidation states simultaneously in sessing the feasibility of storing nuclear environment is a complex, partic- the same solution. Accordingly, the waste in geologic repositories. The Fularly elaborate system. Hundreds light actinides exhibit some of the rich- actinides1 are radioactive, long-lived, of chemically active compounds and est and most involved chemistry in the and highly toxic. Over the last few minerals reside within the earth’s periodic table. decades, vast quantities of transuranic formations, and every patch of rock and As a result, the chemical interactions actinides (those with atomic numbers soil is composed of its own particular of actinides in the environment are in- greater than that of uranium) have been mix. The waters that flow upon and ordinately complex. To predict how an produced inside the fuel rods of com- through these formations harbor a vari- actinide might spread through the envi- mercial nuclear reactors. Currently, ety of organic and inorganic ligands. ronment and how fast that transport most spent fuel rods are stored above- Where the water meets the rock is a might occur, we need to characterize all ground in interim storage facilities, but poorly understood interfacial region that local conditions, including the nature of the plan in the United States and some exhibits its own chemical processes. site-specific minerals, temperature and European countries is to deposit this Likewise, from a chemist’s point of pressure profiles, and the local waters’ nuclear waste in repositories buried view, the actinides are complex pH, redox potential (Eh), and ligand hundreds of meters underground. -

Emergence of Californium As the Second Transitional Element in the Actinide Series
ARTICLE Received 28 Nov 2014 | Accepted 3 Mar 2015 | Published 16 Apr 2015 DOI: 10.1038/ncomms7827 OPEN Emergence of californium as the second transitional element in the actinide series Samantha K. Cary1, Monica Vasiliu2, Ryan E. Baumbach3, Jared T. Stritzinger1, Thomas D. Green1, Kariem Diefenbach1, Justin N. Cross1, Kenneth L. Knappenberger1, Guokui Liu4, Mark A. Silver1, A. Eugene DePrince1, Matthew J. Polinski1, Shelley M. Van Cleve5, Jane H. House1, Naoki Kikugawa6, Andrew Gallagher3, Alexandra A. Arico1, David A. Dixon2 & Thomas E. Albrecht-Schmitt1 A break in periodicity occurs in the actinide series between plutonium and americium as the result of the localization of 5f electrons. The subsequent chemistry of later actinides is thought to closely parallel lanthanides in that bonding is expected to be ionic and complexation should not substantially alter the electronic structure of the metal ions. Here we demonstrate that ligation of californium(III) by a pyridine derivative results in significant deviations in the properties of the resultant complex with respect to that predicted for the free ion. We expand on this by characterizing the americium and curium analogues for comparison, and show that these pronounced effects result from a second transition in periodicity in the actinide series that occurs, in part, because of the stabilization of the divalent oxidation state. The metastability of californium(II) is responsible for many of the unusual properties of californium including the green photoluminescence. 1 Department of Chemistry and Biochemistry, Florida State University, Tallahassee, Florida 32306, USA. 2 Department of Chemistry, The University of Alabama, Tuscaloosa, Alabama 35487, USA. 3 National High Magnetic Field Laboratory, Tallahassee, Florida 32310, USA. -

Crystallochemistry of Actinide Nitrides (U1-Ypuy)N and Effect of Impurities
P2-07 Crystallochemistry of actinide nitrides (U1-yPuy)N and effect of impurities Beauvy M. , Coulon-Picard E., Pelletier M. CEA Cadarache (DEN/DEC) – 13108 Saint-Paul-Lez-Durance Cedex, FRANCE Email : [email protected] Abstract – Investigations on actinide nitrides has been done in our Laboratories for Fast Breeder Reactors since the seventies and some properties are reported to show the interest for these fuels. Today, the actinide nitrides are reconsidered as possible fuels for the future fission reactors (GFR and LMFR selected by the international forum Generation IV). The results of new investigations on crystal structure of mixed mononitrides (U,Pu)N, and the effects of oxygen and carbon contaminations on this structure are presented. The cubic "NaCl-fcc" type structure of actinide nitrides AnN with space group O5/h-Fm3m does not respect the “Vegard law” model for the mixed nitrides (U1-yPuy)N. These nitrides are usually considered with strong metallic character associated with partial ionic bonding, but the ionic contribution in the An-N bonding determined in this work is very important and near 41.6% for UN and PuN. From results published on resistivity of mixed nitrides, the data on bonding must be also modified for partial covalence. This is in good agreement with the experimental lattice parameters which are not compatible with dominant metallic bonding. The numbers of bonding electrons in the nitrides (U1-yPuy)N are revaluated and the low values proposed comparatively with those previously published confirm the strong ionic character with high concentration of An3+ ions. The solubility of oxygen and carbon in actinide nitrides (U1-yPuy)N are discussed from measurements on volumic concentration of actinide oxide phase, total oxygen and carbon contents, and lattice parameter of nitrides. -

Alkali Metal Actinide Complex Halides : Thermochemical and Structural Considerations J
Alkali metal actinide complex halides : thermochemical and structural considerations J. Fuger To cite this version: J. Fuger. Alkali metal actinide complex halides : thermochemical and structural considerations. Journal de Physique Colloques, 1979, 40 (C4), pp.C4-207-C4-213. 10.1051/jphyscol:1979465. jpa- 00218861 HAL Id: jpa-00218861 https://hal.archives-ouvertes.fr/jpa-00218861 Submitted on 1 Jan 1979 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. JOURNAL DE PHYSIQUE Colloque C4, supplément au n° 4, Tome 40, avril 1979, page C4-207 Alkali metal actinide complex halides: thermochemical and structural considerations J. Fuger Institute of Radiochemistry, University of Liege, Sart Tilman, B-4000 Liege, Belgium Résumé. — L'auteur passe en revue l'état actuel de nos connaissances dans le domaine de la thermodynamique des complexes halogènes d'actinides avec les ions alcalins, en portant une attention toute spéciale aux dérivés chlorés et bromes. Lorsque les données thermodynamiques et structurales sont accessibles, il tente de déduire l'évolution de l'énergie de la liaison actinide-halogène au sein d'une série de composés isomorphes ou analogues. Enfin, la variation énergétique au cours de la formation du complexe halogène à partir des halogénures binaires d'actinides et de métaux alcalins est prise pour base en vue de prévoir la stabilité de composés nouveaux, spécialement ceux pour lesquels l'halogénure binaire d'actinide n'a pas été préparé ou est de faible stabilité. -

Institute for Transuranium Elements Karlsruhe
INSTITUTE FOR TRANSURANIUM ELEMENTS KARLSRUHE EUR 5381e A V bri » , ^r-— fa um ÉHEf «f i > ^^^^^· w ^R 1 ^1 ^ Bui of the K a f( s rul le' Λ li c/c'J| **WMÌIIHut e /or Trans uran]Im i F.lcMãmm : Λ i - 4 THE EUROPEAN INSTITUTE FOR TRANSURANIUM ELEMENTS (EURATOM) KARLSRUHE 1975 How to get to the Institute >c, Main Geographischer Verlag Stuttgar rsbach ».^KltAl-í ; åe^JO^"J ' - aeri" 2 4 e S kn l'Airopäisdics lnsiiiiil für Transurane 75 Karlsruhe I. Poitfach 2266 Bundesrepublik Deutschland Telex euru d 07825483 Telefon 07247/841 CONTENTS Preface I. The Institute, its Background and How it Works 7 The Transuranium Elements 7 The European Communities and their Treaties 13 Some Historic Dates 14 Facts and Figures 16 The Management of the Plutonium and Transplutonium Programme 21 Safety First 24 Beyond the Day's Work... 27 The City with the European Touch 29 The European School of Karlsruhe 30 II. Some Results and Achievements 32 Fuel Science and Technology 33 Fuel Preparation and Characterisation 33 Fuel Property Studies 34 Thermodynamics 34 Thermal Properties 36 Phase Diagrams 36 Study of Lattice Defects 37 Diffusion Studies 39 Study of Fuel under Irradiation Design and Execution of Irradiation Experiments 41 Post Irradiation Structural Analysis 42 Simulation Experiments 45 Swelling of Advanced Fuels 45 Corrosion Studies 46 Modelling Work 48 Isotope Analysis 49 Basic Actinide Research 50 Isolation and Purification of Transplutonium Nuclides 50 Preparation of Actinide, Metals and Compounds 51 Chemical and Physical Properties of Actinide Metals and Alloys 52 Selected Bibliography 55 Published by the Commission of the European Communities Directorate-General Scientific and Technical Information and Information Management Luxembourg, 1975 Compiled and edited by H.E.