Mandelic Acid
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Quinic Acid-Mediated Induction of Hypovirulence and a Hypovirulence-Associated Double-Stranded RNA in Rhizoctonia Solani Chunyu Liu
The University of Maine DigitalCommons@UMaine Electronic Theses and Dissertations Fogler Library 8-2001 Quinic Acid-Mediated Induction of Hypovirulence and a Hypovirulence-Associated Double-Stranded RNA in Rhizoctonia Solani Chunyu Liu Follow this and additional works at: http://digitalcommons.library.umaine.edu/etd Part of the Biochemistry Commons, and the Molecular Biology Commons Recommended Citation Liu, Chunyu, "Quinic Acid-Mediated Induction of Hypovirulence and a Hypovirulence-Associated Double-Stranded RNA in Rhizoctonia Solani" (2001). Electronic Theses and Dissertations. 332. http://digitalcommons.library.umaine.edu/etd/332 This Open-Access Dissertation is brought to you for free and open access by DigitalCommons@UMaine. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of DigitalCommons@UMaine. QUlNlC ACID-MEDIATED INDUCTION OF HYPOVIRULENCE AND A HY POVlRULENCE-ASSOCIATED DOUBLESTRANDED RNA IN RHIZOCTONIA SOLANI BY Chunyu Liu B.S. Wuhan University, 1989 MS. Wuhan University. 1992 A THESIS Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy (in Biochemistry and Molecular Biology) The Graduate School The Universrty of Maine August, 2001 Advisory Committee: Stellos Tavantzis, Professor of Plant Pathology, Advisor Seanna Annis, Assistant Professor of Mycology Robert Cashon, Assistant Professor of Biochemistry, Molecular Biology Robert Gundersen, Associate Professor of Biochemistry, Molecular Biology John Singer, Professor of Microbiology QUlNlC ACID-MEDIATED INDUCTION OF HYPOVIRULENCE AND A HYPOVIRULENCE-ASSOCIATED DOUBLE-STRANDED RNA (DSRNA) IN RHIZOCTONIA SOLANI By Chunyu Liu Thesis Advisor: Dr. Stellos Tavantzis An Abstract of the Thesis Presented in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy (in Biochemistry and Molecular Biology) August, 2001 This study is a part of a project focused on the relationship between dsRNA and hypovirulence in R. -

Microbial Degradation of the Morphine Alkaloids Purification and Characterization of Morphine Dehydrogenase from Pseudomonas Putida M10
Biochem. J. (1991) 274, 875-880 (Printed in Great Britain) 875 Microbial degradation of the morphine alkaloids Purification and characterization of morphine dehydrogenase from Pseudomonas putida M10 Neil C. BRUCE,* Clare J. WILMOT, Keith N. JORDAN, Lauren D. Gray STEPHENS and Christopher R. LOWE Institute of Biotechnology, University of Cambridge, Tennis Court Road, Cambridge CB2 1QT, U.K. The NADP+-dependent morphine dehydrogenase that catalyses the oxidation of morphine to morphinone was detected in glucose-grown cells of Pseudomonas putida M 10. A rapid and reliable purification procedure involving two consecutive affinity chromatography steps on immobilized dyes was developed for purifying the enzyme 1216-fold to electrophoretic homogeneity from P. putida M 10. Morphine dehydrogenase was found to be a monomer of Mr 32000 and highly specific with regard to substrates, oxidizing only the C-6 hydroxy group of morphine and codeine. The pH optimum of morphine dehydrogenase was 9.5, and at pH 6.5 in the presence of NADPH the enzyme catalyses the reduction of codeinone to codeine. The Km values for morphine and codeine were 0.46 mm and 0.044 mm respectively. The enzyme was inhibited by thiol-blocking reagents and the metal-complexing reagents 1, 10-phenanthroline and 2,2'-dipyridyl, suggesting that a metal centre may be necessary for activity of the enzyme. INTRODUCTION EXPERIMENTAL The morphine alkaloids have attracted considerable attention Materials owing to their analgaesic properties and, consequently, much Mimetic Orange 3 A6XL and Mimetic Red A6XL were effort in the past has been directed at the production of new obtained from Affinity Chromatography Ltd., Freeport, morphine alkaloids by micro-organisms (lizuka et al., 1960, Ballasalla, Isle of Man, U.K. -

Downloaded As a Text File, Is Completely Dynamic
BMC Bioinformatics BioMed Central Database Open Access ORENZA: a web resource for studying ORphan ENZyme activities Olivier Lespinet and Bernard Labedan* Address: Institut de Génétique et Microbiologie, CNRS UMR 8621, Université Paris-Sud, Bâtiment 400, 91405 Orsay Cedex, France Email: Olivier Lespinet - [email protected]; Bernard Labedan* - [email protected] * Corresponding author Published: 06 October 2006 Received: 25 July 2006 Accepted: 06 October 2006 BMC Bioinformatics 2006, 7:436 doi:10.1186/1471-2105-7-436 This article is available from: http://www.biomedcentral.com/1471-2105/7/436 © 2006 Lespinet and Labedan; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Despite the current availability of several hundreds of thousands of amino acid sequences, more than 36% of the enzyme activities (EC numbers) defined by the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB) are not associated with any amino acid sequence in major public databases. This wide gap separating knowledge of biochemical function and sequence information is found for nearly all classes of enzymes. Thus, there is an urgent need to explore these sequence-less EC numbers, in order to progressively close this gap. Description: We designed ORENZA, a PostgreSQL database of ORphan ENZyme Activities, to collate information about the EC numbers defined by the NC-IUBMB with specific emphasis on orphan enzyme activities. -

Proof of De Novo Synthesis of the Qa Enzymes of Neurospora Crassa During Induction
Proc. Nati. Acad. Sci. USA Vol. 74, No. 10, pp. 4256-4260, October 1977 Biochemistry Proof of de novo synthesis of the qa enzymes of Neurospora crassa during induction [catabolic dehydroquinase (5-dehydroquinate dehydratase)/quinate dehydrogenase/qa gene cluster/eukaryote gene regulation] WILLIAM R. REINERT AND NORMAN H. GILES Program in Genetics, Department of Zoology, University of Georgia, Athens, Georgia 30602 Contributed by Norman H. Giles, July 22, 1977 ABSTRACT In Neurospora crassa three inducible enzymes There is strong genetic evidence that the qa-1 gene encodes a are necessary to catabolize quinic acid to protocatechuic acid. regulatory protein which, in the presence of quinic acid, exerts The three genes encoding these enzymes are tightly linked on chromosome VII near methionine-7 (me-7). This qa cluster in- positive control over the synthesis of the enzymes encoded in cludes a fourth gene, qa-1, which encodes a regulatory protein the three adjacent structural genes (6, 10). The appearance of apparently exerting positive control over transcription of the enzyme activity is very specifically regulated. In the presence other three qa genes. However, an alternative hypothesis is that of a preferred carbon source, e.g., sucrose, the activities of all the qa-I protein simply activates preformed polypeptides de- three enzymes are quite low. Quinate as a sole carbon source rived from the three structural genes. The use of density labeling causes a coordinate induction of all three enzymes (9). This with D20 demonstrated conclusively that the qa enzymes are synthesized de novo only during induction on quinic acid. Na- increase in enzyme activity can be inhibited by the simulta- tive catabolic dehydroquinase (5-dehydroquinate dehydratase; neous addition of cycloheximide to the medium (ref. -

Subcellular Localization of Quinate: Oxidoreductase from Phaseolus Mungo L. Sprouts Xiangbo Kang, H
Subcellular Localization of Quinate: Oxidoreductase from Phaseolus mungo L. Sprouts Xiangbo Kang, H. Ekkehard Neuhaus and Renate Scheibe Pflanzenphysiologie, Fachbereich Biologie/Chemie, Universität Osnabrück, D-49069 Osnabrück, Bundesrepublik Deutschland Z. Naturforsch. 49c, 415-420 (1994); received March 24/April 27, 1994 Phaseolus mungo, Quinate:Oxidoreductase, Quinic Acid, Prechorismate Pathway, Shikimate Pathway Quinate:oxidoreductase (QORase, EC 1.1.1.24) was isolated and purified from etiolated mung bean (Phaseolus mungo L.) sprouts and a monospecific antiserum was raised in rabbit to the homogeneous protein. Highly intact etioplasts were isolated from the same plant material. The stroma of the purified etioplasts was enzymatically characterized. Contami nation by cytosol, mitochondria and vacuole was estimated from activities of marker en zymes. QORase activity was localized in the stroma (about 91% for both NAD+ and NADP+ as a cofactor). Western blotting and immunoprinting of the stroma proteins revealed a single band that migrated identically with the purified QORase. The results suggest that the QOR ase is localized predominantly, if not exclusively, in the etioplast stroma. The physiological role of the enzyme is discussed. Introduction (Refeno et al., 1982), tobacco callus (Beaudoin Quinic acid is widely distributed among higher and Thorpe, 1983, 1984) and conifer needles (Osi and lower plants (Karrer, 1958; Minamikawa and pov and Shein, 1987). Two of them were reported Yoshida, 1972; Boudet, 1973; Yoshida et al, 1975). to be purified to homogeneity (Refeno et al., 1982; Therefore, its physiological function and metab Kang and Scheibe, 1993). However, the character olism are of interest. At the turn of the 1960’s istics of these enzymes appeared to be very diver Weinstein et al. -

All Enzymes in BRENDA™ the Comprehensive Enzyme Information System
All enzymes in BRENDA™ The Comprehensive Enzyme Information System http://www.brenda-enzymes.org/index.php4?page=information/all_enzymes.php4 1.1.1.1 alcohol dehydrogenase 1.1.1.B1 D-arabitol-phosphate dehydrogenase 1.1.1.2 alcohol dehydrogenase (NADP+) 1.1.1.B3 (S)-specific secondary alcohol dehydrogenase 1.1.1.3 homoserine dehydrogenase 1.1.1.B4 (R)-specific secondary alcohol dehydrogenase 1.1.1.4 (R,R)-butanediol dehydrogenase 1.1.1.5 acetoin dehydrogenase 1.1.1.B5 NADP-retinol dehydrogenase 1.1.1.6 glycerol dehydrogenase 1.1.1.7 propanediol-phosphate dehydrogenase 1.1.1.8 glycerol-3-phosphate dehydrogenase (NAD+) 1.1.1.9 D-xylulose reductase 1.1.1.10 L-xylulose reductase 1.1.1.11 D-arabinitol 4-dehydrogenase 1.1.1.12 L-arabinitol 4-dehydrogenase 1.1.1.13 L-arabinitol 2-dehydrogenase 1.1.1.14 L-iditol 2-dehydrogenase 1.1.1.15 D-iditol 2-dehydrogenase 1.1.1.16 galactitol 2-dehydrogenase 1.1.1.17 mannitol-1-phosphate 5-dehydrogenase 1.1.1.18 inositol 2-dehydrogenase 1.1.1.19 glucuronate reductase 1.1.1.20 glucuronolactone reductase 1.1.1.21 aldehyde reductase 1.1.1.22 UDP-glucose 6-dehydrogenase 1.1.1.23 histidinol dehydrogenase 1.1.1.24 quinate dehydrogenase 1.1.1.25 shikimate dehydrogenase 1.1.1.26 glyoxylate reductase 1.1.1.27 L-lactate dehydrogenase 1.1.1.28 D-lactate dehydrogenase 1.1.1.29 glycerate dehydrogenase 1.1.1.30 3-hydroxybutyrate dehydrogenase 1.1.1.31 3-hydroxyisobutyrate dehydrogenase 1.1.1.32 mevaldate reductase 1.1.1.33 mevaldate reductase (NADPH) 1.1.1.34 hydroxymethylglutaryl-CoA reductase (NADPH) 1.1.1.35 3-hydroxyacyl-CoA -
Enzyme Classification
1st Proof 29-7-08 + 0)26-4 ENZYME CLASSIFICATION 2.1 INTRODUCTION 2.1.1 Enzymes Enzymes are biological catalysts that increase the rate of chemical reactions by lowering the activation energy. The molecules involved in the enzyme mediated reactions are known as substrates and the outcome of the reaction or yield is termed product. Generally, the chemical nature of most of the enzymes are proteins and rarely of other types (e.g., RNA). The enzymes are too specific towards their substrates to which they react and thereby the reaction will also be so specific. Sometimes the enzyme needs the presence of a non-protein component (co-enzyme, if it is a vitamin derived organic compound or co-factor, if it is a metal ion) for accomplishing the reaction. In this case, the whole enzyme may be called a holoenzyme, the protein part as apoenzyme and the non-protein constituent a prosthetic group. 2.1.2 Enzyme Nomenclature Principles The sixth complete edition of Enzyme Nomenclature, was published under the patronage of the International Union of Biochemistry and Molecular Biology (formerly the International Union of Biochemistry). By the late 1950s it had become evident that the nomenclature of enzymology was not following the guidelines formulated owing to an increase in the number of enzymes. The naming of enzymes by individual workers had proved far from satisfactory in practice. In many cases the same enzymes were known by several different names, while conversely the same name was sometimes coined to different enzymes. Many of the names conveyed little or no idea about the nature of the reactions catalyzed. -

Subcellular Localization of Quinate: Oxidoreductase from Phaseolus Mungo L. Sprouts Xiangbo Kang, H
Subcellular Localization of Quinate: Oxidoreductase from Phaseolus mungo L. Sprouts Xiangbo Kang, H. Ekkehard Neuhaus and Renate Scheibe Pflanzenphysiologie, Fachbereich Biologie/Chemie, Universität Osnabrück, D-49069 Osnabrück, Bundesrepublik Deutschland Z. Naturforsch. 49c, 415-420 (1994); received March 24/April 27, 1994 Phaseolus mungo, Quinate:Oxidoreductase, Quinic Acid, Prechorismate Pathway, Shikimate Pathway Quinate:oxidoreductase (QORase, EC 1.1.1.24) was isolated and purified from etiolated mung bean (Phaseolus mungo L.) sprouts and a monospecific antiserum was raised in rabbit to the homogeneous protein. Highly intact etioplasts were isolated from the same plant material. The stroma of the purified etioplasts was enzymatically characterized. Contami nation by cytosol, mitochondria and vacuole was estimated from activities of marker en zymes. QORase activity was localized in the stroma (about 91% for both NAD+ and NADP+ as a cofactor). Western blotting and immunoprinting of the stroma proteins revealed a single band that migrated identically with the purified QORase. The results suggest that the QOR ase is localized predominantly, if not exclusively, in the etioplast stroma. The physiological role of the enzyme is discussed. Introduction (Refeno et al., 1982), tobacco callus (Beaudoin Quinic acid is widely distributed among higher and Thorpe, 1983, 1984) and conifer needles (Osi and lower plants (Karrer, 1958; Minamikawa and pov and Shein, 1987). Two of them were reported Yoshida, 1972; Boudet, 1973; Yoshida et al, 1975). to be purified to homogeneity (Refeno et al., 1982; Therefore, its physiological function and metab Kang and Scheibe, 1993). However, the character olism are of interest. At the turn of the 1960’s istics of these enzymes appeared to be very diver Weinstein et al. -

Springer Handbook of Enzymes
Dietmar Schomburg Ida Schomburg (Eds.) Springer Handbook of Enzymes Alphabetical Name Index 1 23 © Springer-Verlag Berlin Heidelberg New York 2010 This work is subject to copyright. All rights reserved, whether in whole or part of the material con- cerned, specifically the right of translation, printing and reprinting, reproduction and storage in data- bases. The publisher cannot assume any legal responsibility for given data. Commercial distribution is only permitted with the publishers written consent. Springer Handbook of Enzymes, Vols. 1–39 + Supplements 1–7, Name Index 2.4.1.60 abequosyltransferase, Vol. 31, p. 468 2.7.1.157 N-acetylgalactosamine kinase, Vol. S2, p. 268 4.2.3.18 abietadiene synthase, Vol. S7,p.276 3.1.6.12 N-acetylgalactosamine-4-sulfatase, Vol. 11, p. 300 1.14.13.93 (+)-abscisic acid 8’-hydroxylase, Vol. S1, p. 602 3.1.6.4 N-acetylgalactosamine-6-sulfatase, Vol. 11, p. 267 1.2.3.14 abscisic-aldehyde oxidase, Vol. S1, p. 176 3.2.1.49 a-N-acetylgalactosaminidase, Vol. 13,p.10 1.2.1.10 acetaldehyde dehydrogenase (acetylating), Vol. 20, 3.2.1.53 b-N-acetylgalactosaminidase, Vol. 13,p.91 p. 115 2.4.99.3 a-N-acetylgalactosaminide a-2,6-sialyltransferase, 3.5.1.63 4-acetamidobutyrate deacetylase, Vol. 14,p.528 Vol. 33,p.335 3.5.1.51 4-acetamidobutyryl-CoA deacetylase, Vol. 14, 2.4.1.147 acetylgalactosaminyl-O-glycosyl-glycoprotein b- p. 482 1,3-N-acetylglucosaminyltransferase, Vol. 32, 3.5.1.29 2-(acetamidomethylene)succinate hydrolase, p. 287 Vol. -

(19) United States (12) Patent Application Publication (10) Pub
US 20130244920A1 (19) United States (12) Patent Application Publication (10) Pub. N0.: US 2013/0244920 A1 Lee et al. (43) Pub. Date: Sep. 19, 2013 (54) WATER SOLUBLE COMPOSITIONS (52) US. Cl. INCORPORATING ENZYMES, AND METHOD USPC ......................................... .. 510/392; 264/299 OF MAKING SAME (57) ABSTRACT (76) Inventors: David M. Lee, CroWn Point, IN (US); Jennifer L‘ Sims’ Lowell’ IN (Us) Disclosed herein are Water soluble compositions, such as ?lms, including a mixture of a ?rst Water-soluble resin, an (21) Appl' NO': 13/422’709 enzyme, and an enzyme stabilizer Which comprises a func (22) Filed: Man 16, 2012 tional substrate for the enzyme, methods of making such compositions, and methods of using such compositions, e.g. Publication Classi?cation to make packets containing functional ingredients. The enzymes can include proteases and mixtures of proteases (51) Int. Cl. With other enzymes, and the compositions provide good C11D 3/386 (2006.01) retention of enzyme function following ?lm processing and B29C 39/02 (2006.01) storage. US 2013/0244920 A1 Sep. 19,2013 WATER SOLUBLE COMPOSITIONS preheated to a temperature less than 77° C., optionally in a INCORPORATING ENZYMES, AND METHOD range ofabout 66° C. to about 77° C., or about 74° C.; drying OF MAKING SAME the Water from the cast mixture over a period of less than 24 hours, optionally less than 12 hours, optionally less than 8 FIELD OF THE DISCLOSURE hours, optionally less than 2 hours, optionally less than 1 [0001] The present disclosure relates generally to Water hour, optionally less than 45 minutes, optionally less than 30 soluble ?lms. -

Structures of Shikimate Dehydrogenase Aroe and Its Paralog Ydib a COMMON STRUCTURAL FRAMEWORK for DIFFERENT ACTIVITIES*
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 278, No. 21, Issue of May 23, pp. 19463–19472, 2003 © 2003 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A. Structures of Shikimate Dehydrogenase AroE and Its Paralog YdiB A COMMON STRUCTURAL FRAMEWORK FOR DIFFERENT ACTIVITIES* Received for publication, January 24, 2003, and in revised form, March 10, 2003 Published, JBC Papers in Press, March 12, 2003, DOI 10.1074/jbc.M300794200 Gurvan Michel‡, Aleksander W. Roszak§¶,Ve´ronique Sauve´‡, John Maclean§, Allan Matte‡, John R. Coggins¶, Miroslaw Cygler‡ʈ, and Adrian J. Lapthorn§** From the ‡Biotechnology Research Institute, NRC Macromolecular Structure Group, Montreal, Quebec H4P 2R2, Canada and the §Department of Chemistry and ¶Division of Biochemistry and Molecular Biology, Institute of Biomedical and Life Sciences, University of Glasgow, Glasgow G12 8QQ, Scotland, United Kingdom Shikimate dehydrogenase catalyzes the fourth step of opment of nontoxic herbicides (3), as well as antimicrobial (4) the shikimate pathway, the essential route for the bio- and antiparasite (2) agents. The sixth step in the pathway, synthesis of aromatic compounds in plants and micro- catalyzed by 5-enolpyruvylshikimate-3-phosphate synthase, organisms. Absent in metazoans, this pathway is an at- has already been successfully targeted, with the development tractive target for nontoxic herbicides and drugs. of glyphosate, a broad spectrum herbicide (5). However, after Escherichia coli expresses two shikimate dehydrogen- 20 years of extensive use, glyphosate-resistant weeds have ase paralogs, the NADP-specific AroE and a putative recently emerged (6), emphasizing the importance of maintain- enzyme YdiB. Here we characterize YdiB as a dual spec- ing target diversity. -
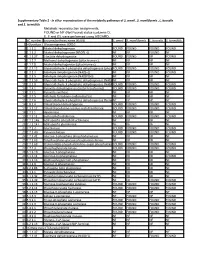
Supplementary Table 2 - in Silico Reconstruction of the Metabolic Pathways of S
Supplementary Table 2 - In silico reconstruction of the metabolic pathways of S. amnii , S. moniliformis , L. buccalis and S. termiditis Metabolic reconstruction assignments, FOUND or NF (Not Found) status (columns D, E, F and G), were performed using ASGARD, EC number Enzyme/pathway name (KEGG) S. amnii S. moniliformis L. buccalis S. termiditis 1 >Glycolysis / Gluconeogenesis 00010 2 1.1.1.1 Alcohol dehydrogenase. FOUND FOUND FOUND FOUND 3 1.1.1.2 Alcohol dehydrogenase (NADP(+)). NF NF FOUND NF 4 1.1.1.27 L-lactate dehydrogenase. FOUND FOUND NF FOUND 5 1.1.2.7 Methanol dehydrogenase (cytochrome c). NF NF NF NF 6 1.1.2.8 Alcohol dehydrogenase (cytochrome c). NF NF NF NF 7 1.2.1.12 Glyceraldehyde-3-phosphate dehydrogenase (phosphorylating).FOUND FOUND FOUND FOUND 8 1.2.1.3 Aldehyde dehydrogenase (NAD(+)). NF NF FOUND FOUND 9 1.2.1.5 Aldehyde dehydrogenase (NAD(P)(+)). NF NF NF NF 10 1.2.1.59 Glyceraldehyde-3-phosphate dehydrogenase (NAD(P)(+))NF (phosphorylating).NF NF NF 11 1.2.1.9 Glyceraldehyde-3-phosphate dehydrogenase (NADP(+)).FOUND FOUND FOUND FOUND 12 1.2.4.1 Pyruvate dehydrogenase (acetyl-transferring). FOUND FOUND FOUND FOUND 13 1.2.7.1 Pyruvate synthase. NF NF NF NF 14 1.2.7.5 Aldehyde ferredoxin oxidoreductase. NF NF NF NF 15 1.2.7.6 Glyceraldehyde-3-phosphate dehydrogenase (ferredoxin).NF NF NF NF 16 1.8.1.4 Dihydrolipoyl dehydrogenase. FOUND FOUND FOUND FOUND 17 2.3.1.12 Dihydrolipoyllysine-residue acetyltransferase. FOUND FOUND FOUND FOUND 18 2.7.1.1 Hexokinase.