Ubiquitin Regulation: the Histone Modifying Enzyme's Story
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Versatile Roles of K63-Linked Ubiquitin Chains in Trafficking
Cells 2014, 3, 1027-1088; doi:10.3390/cells3041027 OPEN ACCESS cells ISSN 2073-4409 www.mdpi.com/journal/cells Review Versatile Roles of K63-Linked Ubiquitin Chains in Trafficking Zoi Erpapazoglou 1,2, Olivier Walker 3 and Rosine Haguenauer-Tsapis 1,* 1 Institut Jacques Monod-CNRS, UMR 7592, Université-Paris Diderot, Sorbonne Paris Cité, F-75205 Paris, France; E-Mail: [email protected] 2 Current address: Brain and Spine Institute, CNRS UMR 7225, Inserm, U 1127, UPMC-P6 UMR S 1127, 75013 Paris, France 3 Institut des Sciences Analytiques, UMR5280, Université de Lyon/Université Lyon 1, 69100 Villeurbanne, France; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]. External Editor: Hanjo Hellmann Received: 14 July 2014; in revised form: 14 October 2014 / Accepted: 21 October 2014 / Published: 12 November 2014 Abstract: Modification by Lys63-linked ubiquitin (UbK63) chains is the second most abundant form of ubiquitylation. In addition to their role in DNA repair or kinase activation, UbK63 chains interfere with multiple steps of intracellular trafficking. UbK63 chains decorate many plasma membrane proteins, providing a signal that is often, but not always, required for their internalization. In yeast, plants, worms and mammals, this same modification appears to be critical for efficient sorting to multivesicular bodies and subsequent lysosomal degradation. UbK63 chains are also one of the modifications involved in various forms of autophagy (mitophagy, xenophagy, or aggrephagy). Here, in the context of trafficking, we report recent structural studies investigating UbK63 chains assembly by various E2/E3 pairs, disassembly by deubiquitylases, and specifically recognition as sorting signals by receptors carrying Ub-binding domains, often acting in tandem. -

Structural Basis for High-Affinity Peptide Inhibition of P53 Interactions with MDM2 and MDMX
Structural basis for high-affinity peptide inhibition of p53 interactions with MDM2 and MDMX Marzena Pazgiera,1, Min Liua,b,1, Guozhang Zoua, Weirong Yuana, Changqing Lia, Chong Lia, Jing Lia, Juahdi Monboa, Davide Zellaa, Sergey G. Tarasovc, and Wuyuan Lua,2 aInstitute of Human Virology, University of Maryland School of Medicine, 725 West Lombard Street, Baltimore, MD 21201; bThe First Affiliated Hospital, School of Medicine, Xi’an Jiaotong University, Shaanxi Province 710061, China; and cStructural Biophysics Laboratory, National Cancer Institute at Frederick, Frederick, MD 21702 Communicated by Robert C. Gallo, University of Maryland, Baltimore, MD, January 28, 2009 (received for review September 29, 2008) The oncoproteins MDM2 and MDMX negatively regulate the ac- ligase activity (10). Structurally related to MDM2, MDMX of tivity and stability of the tumor suppressor protein p53—a cellular 490-aa residues possesses domain structures arranged similarly process initiated by MDM2 and/or MDMX binding to the N- to MDM2, except that MDMX lacks ubiquitin-ligase function terminal transactivation domain of p53. MDM2 and MDMX in many (11, 12). Growing evidence supports that in unstressed cells tumors confer p53 inactivation and tumor survival, and are impor- MDM2 primarily controls p53 stability through ubiquitylation to tant molecular targets for anticancer therapy. We screened a target the tumor suppressor protein for constitutive degradation duodecimal peptide phage library against site-specifically biotin- by the proteasome (13, 14), whereas MDMX mainly functions as ylated p53-binding domains of human MDM2 and MDMX chemi- a significant p53 transcriptional antagonist independently of cally synthesized via native chemical ligation, and identified sev- MDM2 (15, 16). -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

The Ubiquitin Proteasome System and Its Involvement in Cell Death Pathways
Cell Death and Differentiation (2010) 17, 1–3 & 2010 Macmillan Publishers Limited All rights reserved 1350-9047/10 $32.00 www.nature.com/cdd Editorial The ubiquitin proteasome system and its involvement in cell death pathways F Bernassola1, A Ciechanover2 and G Melino1,3 Cell Death and Differentiation (2010) 17, 1–3; doi:10.1038/cdd.2009.189 Following the awarding of the 2004 Nobel Prize in Chemistry Inactivation of the proteasome following caspase-mediated to Aaron Ciechanover, Avram Hershko, and Irwin A Rose cleavage may disable the proteasome, interfering with its for the discovery of ubiquitin (Ub)-mediated degradation, role in the regulation of key cellular processes and thereby Cell Death and Differentiation has drawn the attention of facilitating induction of apoptosis. The noted recent develop- its readers to the Ub Proteasome System (UPS) and its ments show how understanding of these functions is just involvement in regulating cell death pathways.1–4 The current starting to emerge. For example, why does dIAP1 associate set of reviews is an update on this theme.5–16 with multiple E2s via its RING finger? Does dIAP1 also interact From previous review articles published in Cell Death and with the E3 – the F-box protein Morgue, which is a part of an Differentiation, it was apparent that the UPS has a major SCF E3 complex? Why does dIAP1, which is an E3, have to mechanistic role in regulating cell death via modification and interact with other ligases such as the N-end rule UBR1 and degradation of key regulatory proteins involved in -

PGC-1A Protects from Notch-Induced Kidney Fibrosis Development
BASIC RESEARCH www.jasn.org PGC-1a Protects from Notch-Induced Kidney Fibrosis Development † ‡ ‡ Seung Hyeok Han,* Mei-yan Wu, § Bo Young Nam, Jung Tak Park,* Tae-Hyun Yoo,* ‡ † † † † Shin-Wook Kang,* Jihwan Park, Frank Chinga, Szu-Yuan Li, and Katalin Susztak *Department of Internal Medicine, Institute of Kidney Disease Research, Yonsei University College of Medicine, Seoul, Korea; †Renal Electrolyte and Hypertension Division, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania; ‡Severance Biomedical Science Institute, Brain Korea 21 PLUS, Yonsei University College of Medicine, Seoul, Korea; and §Department of Nephrology, The First Hospital of Jilin University, Changchun, China ABSTRACT Kidney fibrosis is the histologic manifestation of CKD. Sustained activation of developmental pathways, such as Notch, in tubule epithelial cells has been shown to have a key role in fibrosis development. The molecular mechanism of Notch-induced fibrosis, however, remains poorly understood. Here, we show that, that expression of peroxisomal proliferation g-coactivator (PGC-1a) and fatty acid oxidation-related genes are lower in mice expressing active Notch1 in tubular epithelial cells (Pax8-rtTA/ICN1) compared to littermate controls. Chromatin immunoprecipitation assays revealed that the Notch target gene Hes1 directly binds to the regulatory region of PGC-1a. Compared with Pax8-rtTA/ICN1 transgenic animals, Pax8-rtTA/ICN1/Ppargc1a transgenic mice showed improvement of renal structural alterations (on his- tology) and molecular defect (expression of profibrotic genes). Overexpression of PGC-1a restored mi- tochondrial content and reversed the fatty acid oxidation defect induced by Notch overexpression in vitro in tubule cells. Furthermore, compared with Pax8-rtTA/ICN1 mice, Pax8-rtTA/ICN1/Ppargc1a mice exhibited improvement in renal fatty acid oxidation gene expression and apoptosis. -

The Involvement of Ubiquitination Machinery in Cell Cycle Regulation and Cancer Progression
International Journal of Molecular Sciences Review The Involvement of Ubiquitination Machinery in Cell Cycle Regulation and Cancer Progression Tingting Zou and Zhenghong Lin * School of Life Sciences, Chongqing University, Chongqing 401331, China; [email protected] * Correspondence: [email protected] Abstract: The cell cycle is a collection of events by which cellular components such as genetic materials and cytoplasmic components are accurately divided into two daughter cells. The cell cycle transition is primarily driven by the activation of cyclin-dependent kinases (CDKs), which activities are regulated by the ubiquitin-mediated proteolysis of key regulators such as cyclins, CDK inhibitors (CKIs), other kinases and phosphatases. Thus, the ubiquitin-proteasome system (UPS) plays a pivotal role in the regulation of the cell cycle progression via recognition, interaction, and ubiquitination or deubiquitination of key proteins. The illegitimate degradation of tumor suppressor or abnormally high accumulation of oncoproteins often results in deregulation of cell proliferation, genomic instability, and cancer occurrence. In this review, we demonstrate the diversity and complexity of the regulation of UPS machinery of the cell cycle. A profound understanding of the ubiquitination machinery will provide new insights into the regulation of the cell cycle transition, cancer treatment, and the development of anti-cancer drugs. Keywords: cell cycle regulation; CDKs; cyclins; CKIs; UPS; E3 ubiquitin ligases; Deubiquitinases (DUBs) Citation: Zou, T.; Lin, Z. The Involvement of Ubiquitination Machinery in Cell Cycle Regulation and Cancer Progression. 1. Introduction Int. J. Mol. Sci. 2021, 22, 5754. https://doi.org/10.3390/ijms22115754 The cell cycle is a ubiquitous, complex, and highly regulated process that is involved in the sequential events during which a cell duplicates its genetic materials, grows, and di- Academic Editors: Kwang-Hyun Bae vides into two daughter cells. -

Targeting the Methyltransferase SETD8 Impairs Tumor Cell Survival and Overcomes Drug Resistance Independently of P53 Status in Multiple Myeloma
bioRxiv preprint doi: https://doi.org/10.1101/776930; this version posted September 20, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Targeting the methyltransferase SETD8 impairs tumor cell survival and overcomes drug resistance independently of p53 status in multiple myeloma Laurie Herviou (1,2,4), Fanny Izard (3,4), Ouissem Karmous-Gadacha (2) , Claire Gourzones (1), Celine Bellanger (1), Eva Desmedt (5), Anqi Ma (6), Laure Vincent (7) , Guillaume Cartron (4,7), Karin Vanderkerken (5), Jian Jin (6), Elke De Bruyne (5), Charlotte Grimaud (3,4,8), Eric Julien (3,4,8 +) and Jérôme Moreaux (1,2,4+) (1) IGH, CNRS, Univ Montpellier, France (2) CHU Montpellier, Laboratory for Monitoring Innovative Therapies, Department of Biological Hematology, Montpellier, France (3) Institut de Recherche en Cancérologie de Montpellier (IRCM), INSERM U1194, Institut Régional du Cancer (ICM), Montpellier F-34298, France (4) University of Montpellier, Montpellier, F-34090, France (5) Department of Hematology and Immunology-Myeloma Center Brussels, Vrije Universiteit Brussel, Brussels, Belgium (6) Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, NeW York, NeW York 10029, United States. (7) CHU Montpellier, Department of Clinical Hematology, Montpellier, France (8) Centre National de la Recherche Scientifique (CNRS), F-34293, Montpellier, France + : co-last and corresponding authors ; corresponding authors: Eric Julien ([email protected]) and jérôme Moreaux ([email protected]). 1 bioRxiv preprint doi: https://doi.org/10.1101/776930; this version posted September 20, 2019. -

TRCP Promotes Cell Growth by Targeting PR-Set7/Set8 for Degradation
SCFβ-TRCP promotes cell growth by targeting PR-Set7/Set8 for degradation The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Wang, Z., X. Dai, J. Zhong, H. Inuzuka, L. Wan, X. Li, L. Wang, et al. 2015. “SCFβ-TRCP promotes cell growth by targeting PR-Set7/Set8 for degradation.” Nature Communications 6 (1): 10185. doi:10.1038/ ncomms10185. http://dx.doi.org/10.1038/ncomms10185. Published Version doi:10.1038/ncomms10185 Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:23993465 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA ARTICLE Received 4 May 2015 | Accepted 12 Nov 2015 | Published 15 Dec 2015 DOI: 10.1038/ncomms10185 OPEN SCFb-TRCP promotes cell growth by targeting PR-Set7/Set8 for degradation Zhiwei Wang1,2,*, Xiangpeng Dai2,*, Jiateng Zhong2,3,*, Hiroyuki Inuzuka2, Lixin Wan2, Xiaoning Li2,4, Lixia Wang1, Xiantao Ye1, Liankun Sun4, Daming Gao2,5,LeeZou6 & Wenyi Wei2 The Set8/PR-Set7/KMT5a methyltransferase plays critical roles in governing transcriptional regulation, cell cycle progression and tumorigenesis. Although CRL4Cdt2 was reported to regulate Set8 stability, deleting the PIP motif only led to partial resistance to ultraviolet- induced degradation of Set8, indicating the existence of additional E3 ligase(s) controlling Set8 stability. Furthermore, it remains largely undefined how DNA damage-induced kinase cascades trigger the timely destruction of Set8 to govern tumorigenesis. -

Folding, Function and Subcellular Localization of Parkin
Dissertation zur Erlangung des Doktorgrades der Fakultät für Chemie und Pharmazie der Ludwig-Maximilians-Universtität München Folding, function and subcellular localization of parkin Julia Schlehe aus München 2008 Erklärung Diese Dissertation wurde im Sinne von §13 Abs. 3 der Promotionsordnung vom 29. Januar 1998 von PD Dr. Winklhofer betreut. Ehrenwörtliche Versicherung Diese Dissertation wurde selbständig, ohne unerlaubte Hilfe erarbeitet. München, am 07.10.2008 …………………………………….. (Julia Schlehe) Dissertation eingereicht am 09.10.2008 1. Gutachter PD Dr. Konstanze Winklhofer 2. Gutachter Prof. Dr. Ulrich Hartl Mündliche Prüfung am 10.11.2008 Summary ...............................................................................................................................................................1 Introduction ..........................................................................................................................................................3 Parkinson’s Disease........................................................................................................................................3 History......................................................................................................................................................3 Clinical characteristics, symptoms and treatment ....................................................................................4 Neuropathological characteristics ............................................................................................................6 -

Mutational Landscape Differences Between Young-Onset and Older-Onset Breast Cancer Patients Nicole E
Mealey et al. BMC Cancer (2020) 20:212 https://doi.org/10.1186/s12885-020-6684-z RESEARCH ARTICLE Open Access Mutational landscape differences between young-onset and older-onset breast cancer patients Nicole E. Mealey1 , Dylan E. O’Sullivan2 , Joy Pader3 , Yibing Ruan3 , Edwin Wang4 , May Lynn Quan1,5,6 and Darren R. Brenner1,3,5* Abstract Background: The incidence of breast cancer among young women (aged ≤40 years) has increased in North America and Europe. Fewer than 10% of cases among young women are attributable to inherited BRCA1 or BRCA2 mutations, suggesting an important role for somatic mutations. This study investigated genomic differences between young- and older-onset breast tumours. Methods: In this study we characterized the mutational landscape of 89 young-onset breast tumours (≤40 years) and examined differences with 949 older-onset tumours (> 40 years) using data from The Cancer Genome Atlas. We examined mutated genes, mutational load, and types of mutations. We used complementary R packages “deconstructSigs” and “SomaticSignatures” to extract mutational signatures. A recursively partitioned mixture model was used to identify whether combinations of mutational signatures were related to age of onset. Results: Older patients had a higher proportion of mutations in PIK3CA, CDH1, and MAP3K1 genes, while young- onset patients had a higher proportion of mutations in GATA3 and CTNNB1. Mutational load was lower for young- onset tumours, and a higher proportion of these mutations were C > A mutations, but a lower proportion were C > T mutations compared to older-onset tumours. The most common mutational signatures identified in both age groups were signatures 1 and 3 from the COSMIC database. -

1 the Functional Interplay Between the HIF Pathway and the Ubiquitin System
Zurich Open Repository and Archive University of Zurich Main Library Strickhofstrasse 39 CH-8057 Zurich www.zora.uzh.ch Year: 2017 The functional interplay between the HIF pathway and the ubiquitin system – more than a one-way road Günter, Julia ; Ruiz-Serrano, Amalia ; Pickel, Christina ; Wenger, Roland H ; Scholz, Carsten C Abstract: The hypoxia inducible factor (HIF) pathway and the ubiquitin system represent major cellular processes that are involved in the regulation of a plethora of cellular signaling pathways and tissue func- tions. The ubiquitin system controls the ubiquitination of proteins, which is the covalent linkage of one or several ubiquitin molecules to specific targets. This ubiquitination is catalyzed by approximately 1000 different E3 ubiquitin ligases and can lead to different effects, depending on the type of internal ubiquitin chain linkage. The best-studied function is the targeting of proteins for proteasomal degradation. The activity of E3 ligases is antagonized by proteins called deubiquitinases (or deubiquitinating enzymes), which negatively regulate ubiquitin chains. This is performed in most cases by the catalytic removal of these chains from the targeted protein. The HIF pathway is regulated in an oxygen-dependent manner by oxygen-sensing hydroxylases. Covalent modification of HIF subunits leads to the recruitment ofan E3 ligase complex via the von Hippel-Lindau (VHL) protein and the subsequent polyubiquitination and proteasomal degradation of HIF subunits, demonstrating the regulation of the HIF pathway by the ubiq- uitin system. This unidirectional effect of an E3 ligase on the HIF pathway is the beststudied example for the interplay between these two important cellular processes. However, additional regulatory mechanisms of the HIF pathway through the ubiquitin system are emerging and, more recently, also the reciprocal regulation of the ubiquitin system through components of the HIF pathway. -
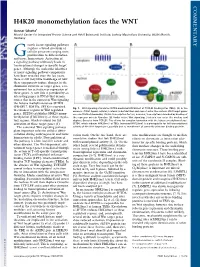
H4K20 Monomethylation Faces the WNT
COMMENTARY H4K20 monomethylation faces the WNT Gunnar Schotta1 Munich Center for Integrated Protein Science and Adolf Butenandt Institute, Ludwig Maximilians University, 80336 Munich, Germany rowth factor signaling pathways regulate a broad spectrum of G cellular processes ranging from proliferation to differentiation and tissue homeostasis. Activation of a signaling pathway ultimately leads to transcriptional changes in specific target genes. Although the molecular identities of many signaling pathway components have been revealed over the last years, there is still very little knowledge of how these components induce changes in the chromatin structure of target genes, a re- quirement for activation or repression of these genes. A new link is provided by an interesting paper in PNAS that demon- strates that in the context of Wnt signaling the histone methyltransferase SETD8 (PR-SET7, KMT5a, SET8) is recruited Fig. 1. Wnt signaling stimulates SETD8-mediated H4K20me1 at TCF/LEF binding sites (TBEs). (A)Inthe to enhancer regions of Wnt-regulated absence of Wnt ligand, cellular β-catenin is destabilized and cannot enter the nucleus. Wnt target genes genes. SETD8 establishes H4K20 mono- are constitutively bound by TCF/LEF transcription factors; however, transcription is blocked by binding of methylation (H4K20me1) at these regula- the repressor protein Groucho. (B) Under active Wnt signaling, β-catenin can enter the nucleus and tory regions, which is crucial for full displace Groucho from TCF/LEF. This allows for complex formation with the histone methyltransferase activation of these target genes (1). SETD8, which induces H4K20me1 at TBEs. Increased H4K20me1 is a prerequisite for full transcriptional The canonical Wnt signaling pathway activity of the Wnt target gene, possibly due to recruitment of currently unknown binding proteins.