Molecular Evolution of NASP and Conserved Histone H3/H4 Transport Pathway Syed Nabeel-Shah1, Kanwal Ashraf2, Ronald E Pearlman2 and Jeffrey Fillingham1*
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

NASP Polyclonal Antibody
For Research Use Only NASP Polyclonal antibody Catalog Number:11323-1-AP Featured Product 7 Publications www.ptgcn.com Catalog Number: GenBank Accession Number: Recommended Dilutions: Basic Information 11323-1-AP BC010105 WB 1:500-1:2400 Size: GeneID (NCBI): IP 0.5-4.0 ug for IP and 1:500-1:2000 700 μg/ml 4678 for WB IHC 1:20-1:200 Source: Full Name: IF 1:10-1:100 Rabbit nuclear autoantigenic sperm protein Isotype: (histone-binding) IgG Calculated MW: Purification Method: 788 aa, 85 kDa Antigen affinity purification Observed MW: Immunogen Catalog Number: 138 kDa AG1824 Applications Tested Applications: Positive Controls: IF, IHC, IP, WB, ELISA WB : mouse testis tissue; Cited Applications: IP : mouse testis tissue; IHC, WB IHC : human testis tissue; human lymphoma tissue Species Specificity: human, mouse, rat IF : MCF-7 cells; Cited Species: human, mouse, rat Note-IHC: suggested antigen retrieval with TE buffer pH 9.0; (*) Alternatively, antigen retrieval may be performed with citrate buffer pH 6.0 NASP (nuclear autoantigenic sperm protein) is associated with DNA replication, cell proliferation and cell cycle Background Information progression through functioning as a Histone H1 binding protein that mediates histone transport to the nucleus. NASP had two isoforms, tNASP (testis type) and sNASP (somatic type). The tNASP was mainly expressed in testis, a variety of malignant tumors, stem cells and embryonic tissues, while sNASP existed in all somatic mitosis cells. This antibody can recognize both isoforms. Notable Publications Author Pubmed ID Journal Application Qing Yuan 25378924 Int J Nanomedicine WB Nishibu Takahiro T 23229118 Biosci Trends WB Sofie De Munter 28032891 FEBS Lett WB Storage: Storage Store at -20ºC. -

Mouse Nasp Conditional Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Nasp Conditional Knockout Project (CRISPR/Cas9) Objective: To create a Nasp conditional knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Nasp gene (NCBI Reference Sequence: NM_016777 ; Ensembl: ENSMUSG00000028693 ) is located on Mouse chromosome 4. 16 exons are identified, with the ATG start codon in exon 2 and the TAA stop codon in exon 16 (Transcript: ENSMUST00000030456). Exon 6~7 will be selected as conditional knockout region (cKO region). Deletion of this region should result in the loss of function of the Mouse Nasp gene. To engineer the targeting vector, homologous arms and cKO region will be generated by PCR using BAC clone RP24-72F14 as template. Cas9, gRNA and targeting vector will be co-injected into fertilized eggs for cKO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Mice homozygous for a null mutation display embryonic lethality before implantation. Exon 6 starts from about 12.94% of the coding region. The knockout of Exon 6~7 will result in frameshift of the gene. The size of intron 5 for 5'-loxP site insertion: 2248 bp, and the size of intron 7 for 3'-loxP site insertion: 4639 bp. The size of effective cKO region: ~2179 bp. The cKO region does not have any other known gene. Page 1 of 8 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele 5' gRNA region gRNA region 3' 1 5 6 7 16 Targeting vector Targeted allele Constitutive KO allele (After Cre recombination) Legends Exon of mouse Nasp Homology arm cKO region loxP site Page 2 of 8 https://www.alphaknockout.com Overview of the Dot Plot Window size: 10 bp Forward Reverse Complement Sequence 12 Note: The sequence of homologous arms and cKO region is aligned with itself to determine if there are tandem repeats. -

Intrinsic Disorder in Tetratricopeptide Repeat Proteins
International Journal of Molecular Sciences Article Intrinsic Disorder in Tetratricopeptide Repeat Proteins 1, 1, 1, 2 Nathan W. Van Bibber y, Cornelia Haerle y, Roy Khalife y, Bin Xue and Vladimir N. Uversky 1,3,4,* 1 Department of Molecular Medicine Morsani College of Medicine, University of South Florida, 12901 Bruce B. Downs Blvd., Tampa, FL 33612, USA; [email protected] (N.W.V.B.); [email protected] (C.H.); [email protected] (R.K.) 2 Department of Cell Biology, Microbiology and Molecular Biology, School of Natural Sciences and Mathematics, College of Arts and Sciences, University of South Florida, Tampa, FL 33620, USA; [email protected] 3 USF Health Byrd Alzheimer’s Research Institute, Morsani College of Medicine, University of South Florida, 12901 Bruce B. Downs Blvd., Tampa, FL 33612, USA 4 Institute for Biological Instrumentation, Russian Academy of Sciences, Federal Research Center “Pushchino Scientific Center for Biological Research of the Russian Academy of Sciences”, 4 Institutskaya St., Pushchino, 142290 Moscow Region, Russia * Correspondence: [email protected]; Tel.: +1-813-974-5816; Fax: +1-813-974-7357 These authors contributed equally to this work. y Received: 21 April 2020; Accepted: 22 May 2020; Published: 25 May 2020 Abstract: Among the realm of repeat containing proteins that commonly serve as “scaffolds” promoting protein-protein interactions, there is a family of proteins containing between 2 and 20 tetratricopeptide repeats (TPRs), which are functional motifs consisting of 34 amino acids. The most distinguishing feature of TPR domains is their ability to stack continuously one upon the other, with these stacked repeats being able to affect interaction with binding partners either sequentially or in combination. -
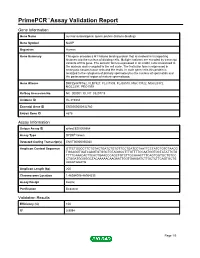
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name nuclear autoantigenic sperm protein (histone-binding) Gene Symbol NASP Organism Human Gene Summary This gene encodes a H1 histone binding protein that is involved in transporting histones into the nucleus of dividing cells. Multiple isoforms are encoded by transcript variants of this gene. The somatic form is expressed in all mitotic cells is localized to the nucleus and is coupled to the cell cycle. The testicular form is expressed in embryonic tissues tumor cells and the testis. In male germ cells this protein is localized to the cytoplasm of primary spermatocytes the nucleus of spermatids and the periacrosomal region of mature spermatozoa. Gene Aliases DKFZp547F162, FLB7527, FLJ31599, FLJ35510, MGC19722, MGC20372, MGC2297, PRO1999 RefSeq Accession No. NC_000001.10, NT_032977.9 UniGene ID Hs.319334 Ensembl Gene ID ENSG00000132780 Entrez Gene ID 4678 Assay Information Unique Assay ID qHsaCED0005564 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000350030 Amplicon Context Sequence GTTCTGGCCTTCTGTACTGATCTGTGTTCCTGATCCTAATTCCTATCTGTCTAACG TGGAGGTGATCAAGTGTGGCTGTAGGCCTTTGTTTTCCAATGGTGCTATATTCTG TTTTCAAACACTTCACTGAACCCAGCTGTCTTGCAAACTTTCAGTGGTGCTGTCC CTGGATGGGGGCTACAAAAACAAGAATTGGTGAAGATCTTGCTCTTCAGTGCTG AAAATGGATG Amplicon Length (bp) 200 Chromosome Location 1:46084006-46084235 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 100 R2 0.9994 Page 1/5 PrimePCR™Assay Validation Report cDNA Cq 20.64 cDNA Tm (Celsius) 83 gDNA Cq 22.63 Specificity -

A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family
Rockefeller University Digital Commons @ RU Student Theses and Dissertations 2018 A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family Linda Molla Follow this and additional works at: https://digitalcommons.rockefeller.edu/ student_theses_and_dissertations Part of the Life Sciences Commons A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY A Thesis Presented to the Faculty of The Rockefeller University in Partial Fulfillment of the Requirements for the degree of Doctor of Philosophy by Linda Molla June 2018 © Copyright by Linda Molla 2018 A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY Linda Molla, Ph.D. The Rockefeller University 2018 APOBEC2 is a member of the AID/APOBEC cytidine deaminase family of proteins. Unlike most of AID/APOBEC, however, APOBEC2’s function remains elusive. Previous research has implicated APOBEC2 in diverse organisms and cellular processes such as muscle biology (in Mus musculus), regeneration (in Danio rerio), and development (in Xenopus laevis). APOBEC2 has also been implicated in cancer. However the enzymatic activity, substrate or physiological target(s) of APOBEC2 are unknown. For this thesis, I have combined Next Generation Sequencing (NGS) techniques with state-of-the-art molecular biology to determine the physiological targets of APOBEC2. Using a cell culture muscle differentiation system, and RNA sequencing (RNA-Seq) by polyA capture, I demonstrated that unlike the AID/APOBEC family member APOBEC1, APOBEC2 is not an RNA editor. Using the same system combined with enhanced Reduced Representation Bisulfite Sequencing (eRRBS) analyses I showed that, unlike the AID/APOBEC family member AID, APOBEC2 does not act as a 5-methyl-C deaminase. -

Novel Targets of Apparently Idiopathic Male Infertility
International Journal of Molecular Sciences Review Molecular Biology of Spermatogenesis: Novel Targets of Apparently Idiopathic Male Infertility Rossella Cannarella * , Rosita A. Condorelli , Laura M. Mongioì, Sandro La Vignera * and Aldo E. Calogero Department of Clinical and Experimental Medicine, University of Catania, 95123 Catania, Italy; [email protected] (R.A.C.); [email protected] (L.M.M.); [email protected] (A.E.C.) * Correspondence: [email protected] (R.C.); [email protected] (S.L.V.) Received: 8 February 2020; Accepted: 2 March 2020; Published: 3 March 2020 Abstract: Male infertility affects half of infertile couples and, currently, a relevant percentage of cases of male infertility is considered as idiopathic. Although the male contribution to human fertilization has traditionally been restricted to sperm DNA, current evidence suggest that a relevant number of sperm transcripts and proteins are involved in acrosome reactions, sperm-oocyte fusion and, once released into the oocyte, embryo growth and development. The aim of this review is to provide updated and comprehensive insight into the molecular biology of spermatogenesis, including evidence on spermatogenetic failure and underlining the role of the sperm-carried molecular factors involved in oocyte fertilization and embryo growth. This represents the first step in the identification of new possible diagnostic and, possibly, therapeutic markers in the field of apparently idiopathic male infertility. Keywords: spermatogenetic failure; embryo growth; male infertility; spermatogenesis; recurrent pregnancy loss; sperm proteome; DNA fragmentation; sperm transcriptome 1. Introduction Infertility is a widespread condition in industrialized countries, affecting up to 15% of couples of childbearing age [1]. It is defined as the inability to achieve conception after 1–2 years of unprotected sexual intercourse [2]. -

The Histone Chaperone Nrp1 Is Required for Chromatin Stability And
Lian et al. Epigenetics & Chromatin (2021) 14:34 https://doi.org/10.1186/s13072-021-00409-4 Epigenetics & Chromatin RESEARCH Open Access The histone chaperone Nrp1 is required for chromatin stability and nuclear division in Tetrahymena thermophila Yinjie Lian1, Huijuan Hao1, Jing Xu1,2, Tao Bo1, Aihua Liang1 and Wei Wang1* Abstract Histone chaperones facilitate DNA replication and repair by promoting chromatin assembly, disassembly and histone exchange. Following histones synthesis and nucleosome assembly, the histones undergo posttranslational modifca- tion by diferent enzymes and are deposited onto chromatins by various histone chaperones. In Tetrahymena ther- mophila, histones from macronucleus (MAC) and micronucleus (MIC) have been comprehensively investigated, but the function of histone chaperones remains unclear. Histone chaperone Nrp1 in Tetrahymena contains four conserved tetratricopepeptide repeat (TPR) domains and one C-terminal nuclear localization signal. TPR2 is typically interrupted by a large acidic motif. Immunofuorescence staining showed that Nrp1 is located in the MAC and MICs, but disap- peared in the apoptotic parental MAC and the degraded MICs during the conjugation stage. Nrp1 was also colocal- ized with α-tubulin around the spindle structure. NRP1 knockdown inhibited cellular proliferation and led to the loss of chromosome, abnormal macronuclear amitosis, and disorganized micronuclear mitosis during the vegetative growth stage. During sexual developmental stage, the gametic nuclei failed to be selected and abnormally degraded in NRP1 knockdown mutants. Afnity purifcation combined with mass spectrometry analysis indicated that Nrp1 is co-purifed with core histones, heat shock proteins, histone chaperones, and DNA damage repair proteins. The physi- cal direct interaction of Nrp1 and Asf1 was also confrmed by pull-down analysis in vitro. -

Centromere Repositioning Causes Inversion of Meiosis and Generates a Reproductive Barrier
Centromere repositioning causes inversion of meiosis and generates a reproductive barrier Min Lua and Xiangwei Hea,1 aMinistry of Education Key Laboratory of Biosystems Homeostasis & Protection and Innovation Center for Cell Signaling Network, Life Sciences Institute, Zhejiang University, 310058 Hangzhou, Zhejiang, China Edited by J. Richard McIntosh, University of Colorado, Boulder, CO, and approved September 20, 2019 (received for review July 10, 2019) The chromosomal position of each centromere is determined postulated that a neocentromere may seed the formation of an epigenetically and is highly stable, whereas incremental cases ENC at a site devoid of satellite DNA, which is then matured have supported the occurrence of centromere repositioning on an through acquisition of repetitive DNA. ENCs and neocentromeres evolutionary time scale (evolutionary new centromeres, ENCs), are considered as two sides of the same coin, manifestations of the which is thought to be important in speciation. The mechanisms same biological phenomenon at drastically different time scales underlying the high stability of centromeres and its functional and population sizes (7). Hence, understanding centromere repo- significance largely remain an enigma. Here, in the fission yeast sitioning may provide mechanistic insights into ENC emergence Schizosaccharomyces pombe, we identify a feedback mechanism: and progression. The kinetochore, whose assembly is guided by the centromere, in CENP-A–containing chromatin directly recruits specific com- turn, enforces centromere stability. Upon going through meiosis, ponents of the kinetochore, called the constitutive centromere- specific inner kinetochore mutations induce centromere reposi- associated network. The kinetochore is a proteinaceous ma- tioning—inactivation of the original centromere and formation chinery comprised of inner and outer parts, each compassing of a new centromere elsewhere—in 1 of the 3 chromosomes at several subcomplexes. -

393LN V 393P 344SQ V 393P Probe Set Entrez Gene
393LN v 393P 344SQ v 393P Entrez fold fold probe set Gene Gene Symbol Gene cluster Gene Title p-value change p-value change chemokine (C-C motif) ligand 21b /// chemokine (C-C motif) ligand 21a /// chemokine (C-C motif) ligand 21c 1419426_s_at 18829 /// Ccl21b /// Ccl2 1 - up 393 LN only (leucine) 0.0047 9.199837 0.45212 6.847887 nuclear factor of activated T-cells, cytoplasmic, calcineurin- 1447085_s_at 18018 Nfatc1 1 - up 393 LN only dependent 1 0.009048 12.065 0.13718 4.81 RIKEN cDNA 1453647_at 78668 9530059J11Rik1 - up 393 LN only 9530059J11 gene 0.002208 5.482897 0.27642 3.45171 transient receptor potential cation channel, subfamily 1457164_at 277328 Trpa1 1 - up 393 LN only A, member 1 0.000111 9.180344 0.01771 3.048114 regulating synaptic membrane 1422809_at 116838 Rims2 1 - up 393 LN only exocytosis 2 0.001891 8.560424 0.13159 2.980501 glial cell line derived neurotrophic factor family receptor alpha 1433716_x_at 14586 Gfra2 1 - up 393 LN only 2 0.006868 30.88736 0.01066 2.811211 1446936_at --- --- 1 - up 393 LN only --- 0.007695 6.373955 0.11733 2.480287 zinc finger protein 1438742_at 320683 Zfp629 1 - up 393 LN only 629 0.002644 5.231855 0.38124 2.377016 phospholipase A2, 1426019_at 18786 Plaa 1 - up 393 LN only activating protein 0.008657 6.2364 0.12336 2.262117 1445314_at 14009 Etv1 1 - up 393 LN only ets variant gene 1 0.007224 3.643646 0.36434 2.01989 ciliary rootlet coiled- 1427338_at 230872 Crocc 1 - up 393 LN only coil, rootletin 0.002482 7.783242 0.49977 1.794171 expressed sequence 1436585_at 99463 BB182297 1 - up 393 -

Durham Research Online
Durham Research Online Deposited in DRO: 09 June 2015 Version of attached le: Published Version Peer-review status of attached le: Peer-reviewed Citation for published item: Schmid, R. and Grellscheid, S.N. and Ehrmann, I. and Dalgliesh, C. and Danilenko, M. and Paronetto, M.P. and Pedrotti, S. and Grellscheid, D. and Dixon, R.J. and Sette, C. and Eperon, I.C. and Elliott, D.J. (2013) 'The splicing landscape is globally reprogrammed during male meiosis.', Nucleic acids research., 41 (22). pp. 10170-10184. Further information on publisher's website: http://dx.doi.org/10.1093/nar/gkt811 Publisher's copyright statement: c The Author(s) 2013. Published by Oxford University Press. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0/), which permits unrestricted reuse, distribution, and reproduction in any medium, provided the original work is properly cited. Additional information: Use policy The full-text may be used and/or reproduced, and given to third parties in any format or medium, without prior permission or charge, for personal research or study, educational, or not-for-prot purposes provided that: • a full bibliographic reference is made to the original source • a link is made to the metadata record in DRO • the full-text is not changed in any way The full-text must not be sold in any format or medium without the formal permission of the copyright holders. Please consult the full DRO policy for further details. Durham University Library, Stockton Road, Durham DH1 3LY, United Kingdom Tel : +44 (0)191 334 3042 | Fax : +44 (0)191 334 2971 https://dro.dur.ac.uk 10170–10184 Nucleic Acids Research, 2013, Vol. -

Table S1. 103 Ferroptosis-Related Genes Retrieved from the Genecards
Table S1. 103 ferroptosis-related genes retrieved from the GeneCards. Gene Symbol Description Category GPX4 Glutathione Peroxidase 4 Protein Coding AIFM2 Apoptosis Inducing Factor Mitochondria Associated 2 Protein Coding TP53 Tumor Protein P53 Protein Coding ACSL4 Acyl-CoA Synthetase Long Chain Family Member 4 Protein Coding SLC7A11 Solute Carrier Family 7 Member 11 Protein Coding VDAC2 Voltage Dependent Anion Channel 2 Protein Coding VDAC3 Voltage Dependent Anion Channel 3 Protein Coding ATG5 Autophagy Related 5 Protein Coding ATG7 Autophagy Related 7 Protein Coding NCOA4 Nuclear Receptor Coactivator 4 Protein Coding HMOX1 Heme Oxygenase 1 Protein Coding SLC3A2 Solute Carrier Family 3 Member 2 Protein Coding ALOX15 Arachidonate 15-Lipoxygenase Protein Coding BECN1 Beclin 1 Protein Coding PRKAA1 Protein Kinase AMP-Activated Catalytic Subunit Alpha 1 Protein Coding SAT1 Spermidine/Spermine N1-Acetyltransferase 1 Protein Coding NF2 Neurofibromin 2 Protein Coding YAP1 Yes1 Associated Transcriptional Regulator Protein Coding FTH1 Ferritin Heavy Chain 1 Protein Coding TF Transferrin Protein Coding TFRC Transferrin Receptor Protein Coding FTL Ferritin Light Chain Protein Coding CYBB Cytochrome B-245 Beta Chain Protein Coding GSS Glutathione Synthetase Protein Coding CP Ceruloplasmin Protein Coding PRNP Prion Protein Protein Coding SLC11A2 Solute Carrier Family 11 Member 2 Protein Coding SLC40A1 Solute Carrier Family 40 Member 1 Protein Coding STEAP3 STEAP3 Metalloreductase Protein Coding ACSL1 Acyl-CoA Synthetase Long Chain Family Member 1 Protein -

Examining the Influence of Histone Chaperones on Nuclear Size
Examining the Influence of Histone Chaperones on Nuclear Size Seth C. Eckhardt Department of Molecular Biology, University of Wyoming, Laramie, WY, 82071 [email protected] Advisor: Dan Levy PhD Abstract Size alterations in the nucleus have been visualized by pathologists for many years to diagnose and prognose cancer. Nuclear size and morphology aberrations are also common in numerous diseases. However, it is unknown whether altered nuclear size is the result or cause of pathogenesis. How the size of the nucleus is regulated is a fundamental cell biological question. A better understanding of the mechanisms that govern nuclear size regulation could elucidate novel therapeutics for cancer and disease. Here we review the structures that constitute the nucleus and delve into some of the mechanisms and components known to be correlated with nuclear size. Some of the regulators that will be covered include nuclear lamins, the endoplasmic reticulum, cell size and ploidy, chromatin condensation and nucleocytoplasmic transport. After discussing each constituent, examples are provided to highlight their implication in various types of cancer. We will then discuss the use of Xenopus laevis egg extracts to illuminate cellular scaling mechanisms and the recent discovery of a histone chaperone as a putative nuclear size effector. This research leads into the implementation of an experimental study to determine if another histone chaperone, nuclear autoantigenic sperm protein, also affects nuclear size. Introduction The nucleus, commonly referred to as the control center of the cell, is the largest organelle. Most eukaryotic cells enclose a single nucleus that surrounds and protects deoxyribonucleic acids (DNA) (Edens et al., 2013).