The Genome of Ectocarpus Subulatus Highlights Unique Mechanisms For
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Sequence from B4 Sponge with (A) the First BLAST Hit Asbestopluma Lycopodium and (B) the Sequence of M
Supplementary Material Figure S1. Alignments of CO1 (PorCOI2fwd/PorCOI2rev) sequence from B4 sponge with (A) the first BLAST hit Asbestopluma lycopodium and (B) the sequence of M. acerata displaying low query cover. 1 Figure S2. Alignment of CO1 (dgLCO1490/dgHCO2198) sequence from B4 sponge with the first BLAST hit (M. acerata). 2 Figure S3. Alignment of CO1 (dgLCO1490/dgHCO2198) sequence from D4 sponge with the first BLAST hit (H. pilosus). 3 Figure S4. Taxonomy Bar Plot, reporting the relative frequencies (in percentage, %) of the bacteria taxons more representative for each of the four sponges under analysis . Sample code: B4= M. (Oxymycale) acerata; D4= H. pilosus, D6= M. sarai, C6= H. (Rhizoniera) dancoi. Each taxon is highlighted by a different color. 4 Figure S5. Krona plot at the seven increasing complexity levels: (a) Regnum, (b) Phylum, (c) Class, (d) Order, (e) Family, (f) Genus and (g) Species. a) 5 b) 6 c) 7 d) 8 e) 9 f) 10 g) 11 Figure S6. Distribution of ASV’s frequencies. 12 Figure S7. Distribution of ASV’s frequencies for each sample (reported as a blue bar). 13 Table S1. BLAST results from B4 sponge (Mycale (Oxymycale) acerata). The primer names, sequence length in base pairs (bp), first hits (highlighted in bold), hits at low significance displaying the correct species (where present), query cover and identity percentages (%) were reported. Sequence Query Identity Primers BLAST results length (bp) cover (%) (%) Mycale macilenta voucher 0CDN7203‐O small subunit 18S A/B 1700 99 98 ribosomal RNA gene, partial sequence Mycale -

Microbial Community Structure and Function on Sinking Particles in the North Pacific Subtropical Gyre
Microbial community structure and function on sinking particles in the North Pacific Subtropical Gyre The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Fontanez, Kristina M., John M. Eppley, Ty J. Samo, David M. Karl, and Edward F. DeLong. “Microbial Community Structure and Function on Sinking Particles in the North Pacific Subtropical Gyre.” Frontiers in Microbiology 6 (May 19, 2015). As Published http://dx.doi.org/10.3389/fmicb.2015.00469 Publisher Frontiers Research Foundation Version Final published version Citable link http://hdl.handle.net/1721.1/98187 Terms of Use Creative Commons Attribution Detailed Terms http://creativecommons.org/licenses/by/4.0/ ORIGINAL RESEARCH published: 19 May 2015 doi: 10.3389/fmicb.2015.00469 Microbial community structure and function on sinking particles in the North Pacific Subtropical Gyre Kristina M. Fontanez 1, John M. Eppley 1, 2, 3, Ty J. Samo 2, 3, 4, David M. Karl 2, 3 and Edward F. DeLong 1, 2, 3* 1 Department of Civil and Environmental Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA, 2 Department of Oceanography, School of Ocean and Earth Science and Technology, University of Hawaii, Honolulu, HI, USA, 3 Daniel K. Inouye Center for Microbial Oceanography: Research and Education, University of Hawaii, Honolulu, HI, USA, 4 Lawrence Livermore National Laboratory, Nuclear and Chemical Sciences Division, Livermore, CA, USA Sinking particles mediate the transport of carbon and energy to the deep-sea, yet the specific microbes associated with sedimenting particles in the ocean’s interior remain largely uncharacterized. In this study, we used particle interceptor traps (PITs) Edited by: to assess the nature of particle-associated microbial communities collected at a variety Anton F. -

Interactions in Self-Assembled Microbial Communities Saturate with Diversity
bioRxiv preprint doi: https://doi.org/10.1101/347948; this version posted June 16, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Interactions in self-assembled microbial communities saturate with diversity Xiaoqian Yu1, Martin F. Polz2*, Eric J. Alm2,3,4,5* 1. Department of Biology, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA 2. Department of Civil and Environmental Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA 3. Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA 4. Center for Microbiome Informatics and Therapeutics, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA 5. Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA *Correspondence should be addressed to: E.J.A. ([email protected]) or M.F.P. ([email protected]) Abstract How the diversity of organisms competing for or sharing resources influences community production is an important question in ecology but has rarely been explored in natural microbial communities. These generally contain large numbers of species making it difficult to disentangle how the effects of different interactions scale with diversity. Here, we show that changing diversity affects measures of community function in relatively simple communities but that increasing richness beyond a threshold has little detectable effect. We generated self-assembled communities with a wide range of diversity by growth of cells from serially diluted seawater on brown algal leachate. We subsequently isolated the most abundant taxa from these communities via dilution-to-extinction in order to compare productivity functions of the entire community to those of individual taxa. -

Genome-Wide Analysis of PL7 Alginate Lyases in the Genus Zobellia
molecules Article Genome-Wide Analysis of PL7 Alginate Lyases in the Genus Zobellia Nadezhda Chernysheva, Evgeniya Bystritskaya, Galina Likhatskaya , Olga Nedashkovskaya and Marina Isaeva * G.B. Elyakov Pacific Institute of Bioorganic Chemistry, Far Eastern Branch, Russian Academy of Sciences, 159, Pr. 100 let Vladivostoku, 690022 Vladivostok, Russia; [email protected] (N.C.); [email protected] (E.B.); [email protected] (G.L.); [email protected] (O.N.) * Correspondence: [email protected]; Tel.: +7-423-231-1168 Abstract: We carried out a detailed investigation of PL7 alginate lyases across the Zobellia genus. The main findings were obtained using the methods of comparative genomics and spatial structure modeling, as well as a phylogenomic approach. Initially, in order to elucidate the alginolytic potential of Zobellia, we calculated the content of polysaccharide lyase (PL) genes in each genome. The genus- specific PLs were PL1, PL6, PL7 (the most abundant), PL14, PL17, and PL40. We revealed that PL7 belongs to subfamilies 3, 5, and 6. They may be involved in local and horizontal gene transfer and gene duplication processes. Most likely, an individual evolution of PL7 genes promotes the genetic variability of the Alginate Utilization System across Zobellia. Apparently, the PL7 alginate lyases may acquire a sub-functionalization due to diversification between in-paralogs. Keywords: Zobellia; genomes; polysaccharide lyase family 7; alginate utilization system; paralogs; orthologs Citation: Chernysheva, N.; Bystritskaya, E.; Likhatskaya, G.; Nedashkovskaya, O.; Isaeva, M. Genome-Wide Analysis of PL7 1. Introduction Alginate Lyases in the Genus Zobellia. Marine algal polysaccharides are an important nutrient source for marine bacteria. To Molecules 2021, 26, 2387. -

The Mannitol Utilization System of the Marine Bacterium Zobellia Galactanivorans Agnès Groisillier, Aurore Labourel, Gurvan Michel, Thierry Tonon
The Mannitol Utilization System of the Marine Bacterium Zobellia galactanivorans Agnès Groisillier, Aurore Labourel, Gurvan Michel, Thierry Tonon To cite this version: Agnès Groisillier, Aurore Labourel, Gurvan Michel, Thierry Tonon. The Mannitol Utilization System of the Marine Bacterium Zobellia galactanivorans. Applied and Environmental Microbiology, Ameri- can Society for Microbiology, 2015, 81, pp.1799 - 1812. 10.1128/AEM.02808-14. hal-01116467 HAL Id: hal-01116467 https://hal.archives-ouvertes.fr/hal-01116467 Submitted on 13 Feb 2015 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. 1 1 The mannitol utilization system of the marine bacterium Zobellia galactanivorans 2 3 4 Agnès Groisillier,a,b, Aurore Labourel,a,b,*, Gurvan Michel,a,b and Thierry Tonona,b,# 5 6 Sorbonne Universités, UPMC Univ Paris 06, UMR 8227, Integrative Biology of Marine 7 Models, Station Biologique de Roscoff, Francea; CNRS, UMR 8227, Integrative Biology of 8 Marine Models, Station Biologique de Roscoff, Franceb 9 10 Running title: Mannitol degradation by Zobellia galactanivorans 11 12 #Address correspondence to Thierry Tonon, [email protected] 13 14 *Present address: University of Newcastle, Cell and Molecular Biosciences, Medical School, 15 Cookson Bidg, Framligton Place, NE2 4HH Newcastle upon Tyne, United Kingdom 16 17 A.G. -
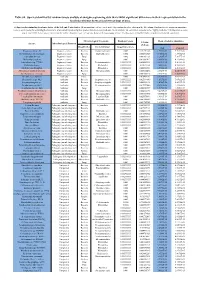
Table S8. Species Identified by Random Forests Analysis of Shotgun Sequencing Data That Exhibit Significant Differences In
Table S8. Species identified by random forests analysis of shotgun sequencing data that exhibit significant differences in their representation in the fecal microbiomes between each two groups of mice. (a) Species discriminating fecal microbiota of the Soil and Control mice. Mean importance of species identified by random forest are shown in the 5th column. Random forests assigns an importance score to each species by estimating the increase in error caused by removing that species from the set of predictors. In our analysis, we considered a species to be “highly predictive” if its importance score was at least 0.001. T-test was performed for the relative abundances of each species between the two groups of mice. P-values were at least 0.05 to be considered statistically significant. Microbiological Taxonomy Random Forests Mean of relative abundance P-Value Species Microbiological Function (T-Test) Classification Bacterial Order Importance Score Soil Control Rhodococcus sp. 2G Engineered strain Bacteria Corynebacteriales 0.002 5.73791E-05 1.9325E-05 9.3737E-06 Herminiimonas arsenitoxidans Engineered strain Bacteria Burkholderiales 0.002 0.005112829 7.1580E-05 1.3995E-05 Aspergillus ibericus Engineered strain Fungi 0.002 0.001061181 9.2368E-05 7.3057E-05 Dichomitus squalens Engineered strain Fungi 0.002 0.018887472 8.0887E-05 4.1254E-05 Acinetobacter sp. TTH0-4 Engineered strain Bacteria Pseudomonadales 0.001333333 0.025523638 2.2311E-05 8.2612E-06 Rhizobium tropici Engineered strain Bacteria Rhizobiales 0.001333333 0.02079554 7.0081E-05 4.2000E-05 Methylocystis bryophila Engineered strain Bacteria Rhizobiales 0.001333333 0.006513543 3.5401E-05 2.2044E-05 Alteromonas naphthalenivorans Engineered strain Bacteria Alteromonadales 0.001 0.000660472 2.0747E-05 4.6463E-05 Saccharomyces cerevisiae Engineered strain Fungi 0.001 0.002980726 3.9901E-05 7.3043E-05 Bacillus phage Belinda Antibiotic Phage 0.002 0.016409765 6.8789E-07 6.0681E-08 Streptomyces sp. -

Transfer of Carbohydrate-Active Enzymes from Marine Bacteria to Japanese Gut Microbiota
Vol 464 | 8 April 2010 | doi:10.1038/nature08937 LETTERS Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota Jan-Hendrik Hehemann1,2{, Gae¨lle Correc1,2, Tristan Barbeyron1,2, William Helbert1,2, Mirjam Czjzek1,2 & Gurvan Michel1,2 Gut microbes supply the human body with energy from dietary not possess the critical residues needed for recognition of agarose or polysaccharides through carbohydrate active enzymes, or k-carrageenan12. CAZymes1, which are absent in the human genome. These enzymes To identify their substrate specificity we cloned and expressed these target polysaccharides from terrestrial plants that dominated diet five GH16 genes in Escherichia coli. However, only Zg1017 and the throughout human evolution2. The array of CAZymes in gut catalytic module of Zg2600 were expressed as soluble proteins and microbes is highly diverse, exemplified by the human gut symbiont could be further analysed (Supplementary Fig. 2). As predicted, these Bacteroides thetaiotaomicron3, which contains 261 glycoside proteins had no activity on commercial agarose (Supplementary hydrolases and polysaccharide lyases, as well as 208 homologues Fig. 3) or k-carrageenan. Consequently, we screened their hydrolytic of susC and susD-genes coding for two outer membrane proteins activity against natural polysaccharides extracted from various marine involved in starch utilization1,4. A fundamental question that, to macrophytes (data not shown). Zg2600 and Zg1017 were found to be our knowledge, has yet to be addressed is how this diversity evolved active only on extracts from the agarophytic red algae Gelidium, by acquiring new genes from microbes living outside the gut. Here Gracilaria and Porphyra, as shown by the release of reducing ends we characterize the first porphyranases from a member of the (Fig. -

Morphogenesis of Ulva Mutabilis (Chlorophyta) Induced by Maribacter Species (Bacteroidetes, Flavobacteriaceae)
Botanica Marina 2017; 60(2): 197–206 Short communication Open Access Anne Weiss, Rodrigo Costa and Thomas Wichard* Morphogenesis of Ulva mutabilis (Chlorophyta) induced by Maribacter species (Bacteroidetes, Flavobacteriaceae) DOI 10.1515/bot-2016-0083 related Flavobacteriaceae and a Maribacter strain isolated Received 31 July, 2016; accepted 11 January, 2017; online first from a red alga did not possess any activity. 17 February, 2017 Keywords: bacteroidetes; cell differentiation; green mac- Abstract: Growth and morphogenesis of the sea lettuce Ulva roalga; morphogens; thallusin. (Chlorophyta) depends on the combination of regulative morphogenetic compounds released by specific associated bacteria. Axenic Ulva gametes develop parthenogenetically The green macroalga Ulva mutabilis Føyn (Chlorophyta) is into callus-like colonies consisting of undifferentiated cells not able to develop and differentiate into blade, stem and without normal cell walls. In Ulva mutabilis Føyn, two bac- rhizoid cells under axenic conditions, or when its microbi- terial strains, Maribacter sp. strain MS6 and Roseovarius ome is not appropriate. Instead, the alga forms callus-like, strain MS2, can restore the complete algal morphogenesis slow growing structures with colourless protrusions from forming a tripartite symbiotic community. Morphogenetic the exterior cell wall (Spoerner et al. 2012, Wichard 2015). compounds ( = morphogens) released by the MS6-strain Early experiments of Provasoli (1958) have already pointed induce rhizoid formation and cell wall development in U. out that treatment of Ulva with antibiotics results in abnor- mutabilis, while several bacteria of the Roseobacter clade, mal growth. Further experiments examined the role of including the MS2-strain, promote blade cell division and isolated bacteria in activating developmental and growth thallus elongation. -

Comparative Genomics and Cazyme Genome Repertoires of Marine Zobellia Amurskyensis KMM 3526T and Zobellia Laminariae KMM 3676T
marine drugs Article Comparative Genomics and CAZyme Genome Repertoires of Marine Zobellia amurskyensis KMM 3526T and Zobellia laminariae KMM 3676T Nadezhda Chernysheva 1, Evgeniya Bystritskaya 1, Anna Stenkova 2, Ilya Golovkin 2, Olga Nedashkovskaya 1 and Marina Isaeva 1,* 1 G.B. Elyakov Pacific Institute of Bioorganic Chemistry, Far Eastern Branch, Russian Academy of Sciences, 159, Pr. 100 let Vladivostoku, Vladivostok 690022, Russia; [email protected] (N.C.); [email protected] (E.B.); [email protected] (O.N.) 2 Far Eastern Federal University, 8 Sukhanova St., Vladivostok 690090, Russia; [email protected] (A.S.); [email protected] (I.G.) * Correspondence: [email protected]; Tel.: +7-914-702-0915 Received: 31 October 2019; Accepted: 22 November 2019; Published: 24 November 2019 Abstract: We obtained two novel draft genomes of type Zobellia strains with estimated genome sizes of 5.14 Mb for Z. amurskyensis KMM 3526Т and 5.16 Mb for Z. laminariae KMM 3676Т. Comparative genomic analysis has been carried out between obtained and known genomes of Zobellia representatives. The pan-genome of Zobellia genus is composed of 4853 orthologous clusters and the core genome was estimated at 2963 clusters. The genus CAZome was represented by 775 GHs classified into 62 families, 297 GTs of 16 families, 100 PLs of 13 families, 112 CEs of 13 families, 186 CBMs of 18 families and 42 AAs of six families. A closer inspection of the carbohydrate-active enzyme (CAZyme) genomic repertoires revealed members of new putative subfamilies of GH16 and GH117, which can be biotechnologically promising for production of oligosaccharides and rare monomers with different bioactivities. -

Carbohydrate Catabolic Capability of a Flavobacteriia Bacterium Isolated
Systematic and Applied Microbiology 42 (2019) 263–274 Contents lists available at ScienceDirect Systematic and Applied Microbiology jou rnal homepage: http://www.elsevier.com/locate/syapm Carbohydrate catabolic capability of a Flavobacteriia bacterium isolated from hadal water a,b,1 a,1 a a a Jiwen Liu , Chun-Xu Xue , Hao Sun , Yanfen Zheng , Zhe Meng , a,b,∗ Xiao-Hua Zhang a MOE Key Laboratory of Marine Genetics and Breeding, College of Marine Life Sciences, Ocean University of China, 5 Yushan Road, Qingdao 266003, China b Laboratory for Marine Ecology and Environmental Science, Qingdao National Laboratory for Marine Science and Technology, Qingdao 266071, China a r t i c l e i n f o a b s t r a c t Article history: Flavobacteriia are abundant in many marine environments including hadal waters, as demonstrated Received 29 September 2018 recently. However, it is unclear how this flavobacterial population adapts to hadal conditions. In this Received in revised form study, extensive comparative genomic analyses were performed for the flavobacterial strain Euzebyella 17 December 2018 marina RN62 isolated from the Mariana Trench hadal water in low abundance. The complete genome of Accepted 15 January 2019 RN62 possessed a considerable number of carbohydrate-active enzymes with a different composition. There was a predominance of GH family 13 proteins compared to closely related relatives, suggesting Keywords: that RN62 has preserved a certain capacity for carbohydrate utilization and that the hadal ocean may Flavobacteriia hold an organic matter reservoir distinct from the surface ocean. Additionally, RN62 possessed potential Hadal water intracellular cycling of the glycogen/starch pathway, which may serve as a strategy for carbon storage Carbohydrate catabolism Organic matter and consumption in response to nutrient pulse and starvation. -

Zobellia Galactanovorans Gen. Nov., Sp. Nov., A
International Journal of Systematic and Evolutionary Microbiology (2001), 51, 985–997 Printed in Great Britain Zobellia galactanovorans gen. nov., sp. nov., a marine species of Flavobacteriaceae isolated from a red alga, and classification of [Cytophaga] uliginosa (ZoBell and Upham 1944) Reichenbach 1989 as Zobellia uliginosa gen. nov., comb. nov. 1 Station Biologique de Tristan Barbeyron,1 Ste! phane L’Haridon,2† Erwan Corre,2 Roscoff, UMR 1931 (CNRS 1 1 and Laboratoire Goe$ mar), Bernard Kloareg and Philippe Potin Place Georges Teissier, 29682 Roscoff Cedex, Bretagne, France Author for correspondence: Tristan Barbeyron. Tel: j33 298 29 23 32. Fax: j33 298 29 23 24. e-mail: barboun!sb-roscoff.fr 2 Station Biologique de Roscoff, UPR 9042 CNRS, Place Georges Teissier, 29680 Roscoff, Bretagne, A mesophilic, aerobic, non-flagellated, gliding bacterium, forming yellow France colonies and designated DsijT, was isolated from a red alga on the sea-shore of Roscoff, Brittany, France. DsijT was selected for its ability to actively degrade both agars and carrageenans. The Gram-negative cells occurred singly or in pairs as long rods. The temperature range for growth was 13–45 SC, with an optimum at 35 SC. The pH range for growth at 35 SC was from 60to85, with an optimum around pH 70. The NaCl concentrations required for growth at 35 SC and pH 70 ranged from 5 to 60 g lN1, with an optimum around 25 g lN1. The GMC content of the genomic DNA was 42–43 mol%. Phylogenetic analysis of 16S rRNA gene sequences indicated that strain DsijT is closely related to [Cytophaga] uliginosa DSM 2061T. -

Marine Bacteroidetes: Distribution Patterns and Role in the Degradation of Organic Matter
Marine Bacteroidetes: distribution patterns and role in the degradation of organic matter Dissertation zur Erlangung des Grades eines Doktors der Naturwissenschaften - Dr. rer. nat. - dem Fachbereich Biologie/Chemie der Universit¨at Bremen vorgelegt von Paola Rocio G´omez Pereira Bremen, Februar 2010 Die vorliegende Arbeit wurde in der Zeit von April 2007 bis Februar 2010 am Max–Planck–Institut f¨ur marine Mikrobiologie in Bremen angefertigt. 1. Gutachter: Prof. Dr. Rudolf Amann 2. Gutachter: Prof. Dr. Victor Smetacek 1. Pr¨ufer: Dr. Bernhard Fuchs 2. Pr¨ufer: Prof. Dr. Ulrich Fischer Tag des Promotionskolloquiums: 9 April 2010 Para mis padres Abstract Oceans occupy two thirds of the Earth’s surface, have a key role in biogeochem- ical cycles, and hold a vast biodiversity. Microorganisms in the world oceans are extremely abundant, their abundance is estimated to be 1029. They have a central role in the recycling of organic matter, therefore they influence the air–sea exchange of carbon dioxide, carbon flux through the food web, and carbon sedimentation by sinking of dead material. Bacteroidetes is one of the most abundant bacterial phyla in marine systems and its members are hypothesized to play a pivotal role in the recycling of organic matter. However, most of the evidence about their role is derived from cultivated species. Bacteroidetes is a highly diverse phylum and cultured strains represent the minority of the marine bacteroidetal community, hence, our knowledge about their ecological role is largely incomplete. In this thesis Bacteroidetes in open ocean and in coastal seas were investigated by a suite of molecular methods.