Genome-Wide Analysis of PL7 Alginate Lyases in the Genus Zobellia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Comparative Proteomic Profiling of Newly Acquired, Virulent And
www.nature.com/scientificreports OPEN Comparative proteomic profling of newly acquired, virulent and attenuated Neoparamoeba perurans proteins associated with amoebic gill disease Kerrie Ní Dhufaigh1*, Eugene Dillon2, Natasha Botwright3, Anita Talbot1, Ian O’Connor1, Eugene MacCarthy1 & Orla Slattery4 The causative agent of amoebic gill disease, Neoparamoeba perurans is reported to lose virulence during prolonged in vitro maintenance. In this study, the impact of prolonged culture on N. perurans virulence and its proteome was investigated. Two isolates, attenuated and virulent, had their virulence assessed in an experimental trial using Atlantic salmon smolts and their bacterial community composition was evaluated by 16S rRNA Illumina MiSeq sequencing. Soluble proteins were isolated from three isolates: a newly acquired, virulent and attenuated N. perurans culture. Proteins were analysed using two-dimensional electrophoresis coupled with liquid chromatography tandem mass spectrometry (LC–MS/MS). The challenge trial using naïve smolts confrmed a loss in virulence in the attenuated N. perurans culture. A greater diversity of bacterial communities was found in the microbiome of the virulent isolate in contrast to a reduction in microbial community richness in the attenuated microbiome. A collated proteome database of N. perurans, Amoebozoa and four bacterial genera resulted in 24 proteins diferentially expressed between the three cultures. The present LC–MS/ MS results indicate protein synthesis, oxidative stress and immunomodulation are upregulated in a newly acquired N. perurans culture and future studies may exploit these protein identifcations for therapeutic purposes in infected farmed fsh. Neoparamoeba perurans is an ectoparasitic protozoan responsible for the hyperplastic gill infection of marine cultured fnfsh referred to as amoebic gill disease (AGD)1. -

Sequence from B4 Sponge with (A) the First BLAST Hit Asbestopluma Lycopodium and (B) the Sequence of M
Supplementary Material Figure S1. Alignments of CO1 (PorCOI2fwd/PorCOI2rev) sequence from B4 sponge with (A) the first BLAST hit Asbestopluma lycopodium and (B) the sequence of M. acerata displaying low query cover. 1 Figure S2. Alignment of CO1 (dgLCO1490/dgHCO2198) sequence from B4 sponge with the first BLAST hit (M. acerata). 2 Figure S3. Alignment of CO1 (dgLCO1490/dgHCO2198) sequence from D4 sponge with the first BLAST hit (H. pilosus). 3 Figure S4. Taxonomy Bar Plot, reporting the relative frequencies (in percentage, %) of the bacteria taxons more representative for each of the four sponges under analysis . Sample code: B4= M. (Oxymycale) acerata; D4= H. pilosus, D6= M. sarai, C6= H. (Rhizoniera) dancoi. Each taxon is highlighted by a different color. 4 Figure S5. Krona plot at the seven increasing complexity levels: (a) Regnum, (b) Phylum, (c) Class, (d) Order, (e) Family, (f) Genus and (g) Species. a) 5 b) 6 c) 7 d) 8 e) 9 f) 10 g) 11 Figure S6. Distribution of ASV’s frequencies. 12 Figure S7. Distribution of ASV’s frequencies for each sample (reported as a blue bar). 13 Table S1. BLAST results from B4 sponge (Mycale (Oxymycale) acerata). The primer names, sequence length in base pairs (bp), first hits (highlighted in bold), hits at low significance displaying the correct species (where present), query cover and identity percentages (%) were reported. Sequence Query Identity Primers BLAST results length (bp) cover (%) (%) Mycale macilenta voucher 0CDN7203‐O small subunit 18S A/B 1700 99 98 ribosomal RNA gene, partial sequence Mycale -

Microbial Community Structure and Function on Sinking Particles in the North Pacific Subtropical Gyre
Microbial community structure and function on sinking particles in the North Pacific Subtropical Gyre The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Fontanez, Kristina M., John M. Eppley, Ty J. Samo, David M. Karl, and Edward F. DeLong. “Microbial Community Structure and Function on Sinking Particles in the North Pacific Subtropical Gyre.” Frontiers in Microbiology 6 (May 19, 2015). As Published http://dx.doi.org/10.3389/fmicb.2015.00469 Publisher Frontiers Research Foundation Version Final published version Citable link http://hdl.handle.net/1721.1/98187 Terms of Use Creative Commons Attribution Detailed Terms http://creativecommons.org/licenses/by/4.0/ ORIGINAL RESEARCH published: 19 May 2015 doi: 10.3389/fmicb.2015.00469 Microbial community structure and function on sinking particles in the North Pacific Subtropical Gyre Kristina M. Fontanez 1, John M. Eppley 1, 2, 3, Ty J. Samo 2, 3, 4, David M. Karl 2, 3 and Edward F. DeLong 1, 2, 3* 1 Department of Civil and Environmental Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA, 2 Department of Oceanography, School of Ocean and Earth Science and Technology, University of Hawaii, Honolulu, HI, USA, 3 Daniel K. Inouye Center for Microbial Oceanography: Research and Education, University of Hawaii, Honolulu, HI, USA, 4 Lawrence Livermore National Laboratory, Nuclear and Chemical Sciences Division, Livermore, CA, USA Sinking particles mediate the transport of carbon and energy to the deep-sea, yet the specific microbes associated with sedimenting particles in the ocean’s interior remain largely uncharacterized. In this study, we used particle interceptor traps (PITs) Edited by: to assess the nature of particle-associated microbial communities collected at a variety Anton F. -

Succession and Cazyme Expression of Marine Bacterial
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.08.416354; this version posted December 8, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Sweet and magnetic: Succession and CAZyme expression of marine bacterial communities encountering a mix of alginate and pectin particles Carina Bunse1,2,*, Hanna Koch3,4,*, Sven Breider3, Meinhard Simon1,3 and Matthias Wietz2,3# 1Helmholtz Institute for Functional Marine Biodiversity at the University of Oldenburg, Germany 2Alfred Wegener Institute Helmholtz Centre for Polar and Marine Research, Germany 3Institute for Chemistry and Biology of the Marine Environment, University of Oldenburg, Germany 4Department of Microbiology, Radboud University Nijmegen, The Netherlands *These authors contributed equally to this work. #Corresponding author HGF-MPG Joint Research Group for Deep-Sea Ecology and Technology Alfred Wegener Institute Helmholtz Centre for Polar and Marine Research Am Handelshafen 12 27570 Bremerhaven Germany Email: [email protected] Telephone +49-471-48311454 bioRxiv preprint doi: https://doi.org/10.1101/2020.12.08.416354; this version posted December 8, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. ABSTRACT Polysaccharide particles are an important nutrient source and microhabitat for marine bacteria. However, substrate-specific bacterial dynamics in a mixture of particle types with different polysaccharide composition, as likely occurring in natural habitats, are undescribed. -

Gene Expression Analysis of Zobellia Galactanivorans During The
ORIGINAL RESEARCH published: 21 September 2017 doi: 10.3389/fmicb.2017.01808 Gene Expression Analysis of Zobellia galactanivorans during the Degradation of Algal Polysaccharides Reveals both Substrate-Specific and Shared Transcriptome-Wide Responses François Thomas 1*, Philippe Bordron 2, 3, 4, Damien Eveillard 5 and Gurvan Michel 1* 1 Sorbonne Universités, UPMC Univ Paris 06, Centre National de la Recherche Scientifique, UMR 8227, Integrative Biology of Marine Models, Station Biologique de Roscoff, Roscoff, France, 2 Sorbonne Universités, UPMC Univ Paris 06, Centre National de la Recherche Scientifique, FR2424, Analysis and Bioinformatics for Marine Science, Station Biologique de 3 4 Edited by: Roscoff, Roscoff, France, Mathomics, Center for Mathematical Modeling, Universidad de Chile, Santiago, Chile, Center for 5 Catherine Ayn Brissette, Genome Regulation (Fondap 15090007), Universidad de Chile, Santiago, Chile, Université de Nantes, Laboratoire des University of North Dakota, Sciences du Numérique de Nantes, Centre National de la Recherche Scientifique, ECN, IMTA, Nantes, France United States Reviewed by: Flavobacteriia are recognized as key players in the marine carbon cycle, due to their Thomas Schweder, ability to efficiently degrade algal polysaccharides both in the open ocean and in coastal University of Greifswald, Germany Matthias Wietz, regions. The chemical complexity of algal polysaccharides, their differences between University of Oldenburg, Germany algal groups and variations through time and space, imply that marine flavobacteria *Correspondence: have evolved dedicated degradation mechanisms and regulation of their metabolism François Thomas [email protected] during interactions with algae. In the present study, we report the first transcriptome-wide Gurvan Michel gene expression analysis for an alga-associated flavobacterium during polysaccharide [email protected] degradation. -

Interactions in Self-Assembled Microbial Communities Saturate with Diversity
bioRxiv preprint doi: https://doi.org/10.1101/347948; this version posted June 16, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Interactions in self-assembled microbial communities saturate with diversity Xiaoqian Yu1, Martin F. Polz2*, Eric J. Alm2,3,4,5* 1. Department of Biology, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA 2. Department of Civil and Environmental Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA 3. Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA 4. Center for Microbiome Informatics and Therapeutics, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA 5. Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA *Correspondence should be addressed to: E.J.A. ([email protected]) or M.F.P. ([email protected]) Abstract How the diversity of organisms competing for or sharing resources influences community production is an important question in ecology but has rarely been explored in natural microbial communities. These generally contain large numbers of species making it difficult to disentangle how the effects of different interactions scale with diversity. Here, we show that changing diversity affects measures of community function in relatively simple communities but that increasing richness beyond a threshold has little detectable effect. We generated self-assembled communities with a wide range of diversity by growth of cells from serially diluted seawater on brown algal leachate. We subsequently isolated the most abundant taxa from these communities via dilution-to-extinction in order to compare productivity functions of the entire community to those of individual taxa. -

Labile Dissolved Organic Matter Compound Characteristics Select
http://www.diva-portal.org This is the published version of a paper published in Frontiers in Microbiology. Citation for the original published paper (version of record): Pontiller, B., Martinez-Garcia, S., Lundin, D., Pinhassi, J. (2020) Labile Dissolved Organic Matter Compound Characteristics Select for Divergence in Marine Bacterial Activity and Transcription Frontiers in Microbiology, 11: 1-19 https://doi.org/10.3389/fmicb.2020.588778 Access to the published version may require subscription. N.B. When citing this work, cite the original published paper. Permanent link to this version: http://urn.kb.se/resolve?urn=urn:nbn:se:lnu:diva-98814 fmicb-11-588778 September 24, 2020 Time: 19:52 # 1 ORIGINAL RESEARCH published: 25 September 2020 doi: 10.3389/fmicb.2020.588778 Labile Dissolved Organic Matter Compound Characteristics Select for Divergence in Marine Bacterial Activity and Transcription Benjamin Pontiller1, Sandra Martínez-García2, Daniel Lundin1 and Jarone Pinhassi1* 1 Centre for Ecology and Evolution in Microbial Model Systems, Linnaeus University, Kalmar, Sweden, 2 Departamento de Ecoloxía e Bioloxía Animal, Universidade de Vigo, Vigo, Spain Bacteria play a key role in the planetary carbon cycle partly because they rapidly assimilate labile dissolved organic matter (DOM) in the ocean. However, knowledge of the molecular mechanisms at work when bacterioplankton metabolize distinct components of the DOM pool is still limited. We, therefore, conducted seawater culture enrichment experiments with ecologically relevant DOM, combining both polymer and monomer model compounds for distinct compound classes. This included carbohydrates (polysaccharides vs. monosaccharides), proteins (polypeptides vs. amino acids), and nucleic acids (DNA vs. nucleotides). We noted pronounced Edited by: changes in bacterial growth, activity, and transcription related to DOM characteristics. -

A Robust Metaproteomics Pipeline for a Holistic Taxonomic and Functional
bioRxiv preprint doi: https://doi.org/10.1101/667428; this version posted June 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 2 Title: A robust metaproteomics pipeline for a holistic taxonomic and functional 3 characterization of microbial communities from marine particles 4 Authors: Doreen Schultz,1 Daniela Zühlke,1 Jörg Bernhardt,1 Thomas Ben Francis,2 Dirk 5 Albrecht,1 Claudia Hirschfeld,1 Stephanie Markert,3 and Katharina Riedel 1* 6 7 Addresses: 1 Institute of Microbiology, University of Greifswald, Greifswald, Germany. 8 2 Max-Planck Institute for Marine Microbiology, Bremen, Germany. 9 3 Institute of Pharmacy, University of Greifswald, Greifswald, Germany. 10 11 *Corresponding author mailing address: Institute of Microbiology, Center for Functional 12 Genomics of Microbes (C_FunGene), University of Greifswald, Felix-Hausdorff-Straße 8, DE- 13 17487 Greifswald, Germany. E-mail [email protected]; Tel (+49) 3834 420 5900 14 15 Running title: Metaproteomic pipeline to analyse marine particles 1 bioRxiv preprint doi: https://doi.org/10.1101/667428; this version posted June 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 16 Originality-Significance Statement 17 Marine particles consist of organic particulate matter (e.g. phyto- or zooplankton) and 18 particle-associated (PA) microbial communities, which are often embedded in a sugary 19 matrix. A significant fraction of the decaying algal biomass in marine ecosystems is expected 20 to be mineralized by PA heterotrophic communities, which are thus greatly contributing to 21 large-scale carbon fluxes. -

The Mannitol Utilization System of the Marine Bacterium Zobellia Galactanivorans Agnès Groisillier, Aurore Labourel, Gurvan Michel, Thierry Tonon
The Mannitol Utilization System of the Marine Bacterium Zobellia galactanivorans Agnès Groisillier, Aurore Labourel, Gurvan Michel, Thierry Tonon To cite this version: Agnès Groisillier, Aurore Labourel, Gurvan Michel, Thierry Tonon. The Mannitol Utilization System of the Marine Bacterium Zobellia galactanivorans. Applied and Environmental Microbiology, Ameri- can Society for Microbiology, 2015, 81, pp.1799 - 1812. 10.1128/AEM.02808-14. hal-01116467 HAL Id: hal-01116467 https://hal.archives-ouvertes.fr/hal-01116467 Submitted on 13 Feb 2015 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. 1 1 The mannitol utilization system of the marine bacterium Zobellia galactanivorans 2 3 4 Agnès Groisillier,a,b, Aurore Labourel,a,b,*, Gurvan Michel,a,b and Thierry Tonona,b,# 5 6 Sorbonne Universités, UPMC Univ Paris 06, UMR 8227, Integrative Biology of Marine 7 Models, Station Biologique de Roscoff, Francea; CNRS, UMR 8227, Integrative Biology of 8 Marine Models, Station Biologique de Roscoff, Franceb 9 10 Running title: Mannitol degradation by Zobellia galactanivorans 11 12 #Address correspondence to Thierry Tonon, [email protected] 13 14 *Present address: University of Newcastle, Cell and Molecular Biosciences, Medical School, 15 Cookson Bidg, Framligton Place, NE2 4HH Newcastle upon Tyne, United Kingdom 16 17 A.G. -

Thèse De Doctorat En Microbiologie
N° d’ordre: 12/2008 – D/ S.N REPUBLIQUE ALGERIENNE DEMOCRATIQUE ET POPULAIRE MINISTERE DE L’ENSEIGNEMENT SUPERIEUR ET DE LA RECHERCHE SCIENTIFIQUE Université des Sciences et de la Technologie HO U A RI BO U M E DIENE Faculté des Sciences Biologiques T H E S E Présentée pour l’obtention du diplôme de D O C T O R A T EN: SCIE N CES DE LA NATURE Spécialité Microbiologie Par M me BEN M A LEK Yamina Sujet Etude de la résistance aux agents antimicrobiens (métaux lourds et antibiotiques) Et évaluation du pouvoir dépolluant chez un nouveau taxon de la famille des Flavobacteriaceae Soutenue le 20/12/2008, devant le jury co mposé de M m e L A R A B A -DJE B A RI Fatima Professeur (FSB/US T H B Alger) Présidente M r H A C E N E Hocine Professeur (FSB/US T H B Alger) Directeur de thèse M m e T O UIL B O U K O F F A Chafia Professeur (FSB/US T H B Alger) Exa minatrice M r B A K O U R Rabah Professeur (FSB/US T H B Alger) Exa minateur M r B E N S O L T A N E Ahm ed Professeur (Univ. O R A N) Exa minateur Résu mé R E S U M E Dans cette étude, nous avons isolé 17 souches bactériennes de différentes niches écologiques polluées principalement par des hydrocarbures. Elles sont à Gram positif, aérobies strictes, im mobiles, non sporulées, pigmentées surtout en jaune avec une morphologie variable. -
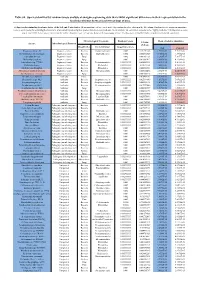
Table S8. Species Identified by Random Forests Analysis of Shotgun Sequencing Data That Exhibit Significant Differences In
Table S8. Species identified by random forests analysis of shotgun sequencing data that exhibit significant differences in their representation in the fecal microbiomes between each two groups of mice. (a) Species discriminating fecal microbiota of the Soil and Control mice. Mean importance of species identified by random forest are shown in the 5th column. Random forests assigns an importance score to each species by estimating the increase in error caused by removing that species from the set of predictors. In our analysis, we considered a species to be “highly predictive” if its importance score was at least 0.001. T-test was performed for the relative abundances of each species between the two groups of mice. P-values were at least 0.05 to be considered statistically significant. Microbiological Taxonomy Random Forests Mean of relative abundance P-Value Species Microbiological Function (T-Test) Classification Bacterial Order Importance Score Soil Control Rhodococcus sp. 2G Engineered strain Bacteria Corynebacteriales 0.002 5.73791E-05 1.9325E-05 9.3737E-06 Herminiimonas arsenitoxidans Engineered strain Bacteria Burkholderiales 0.002 0.005112829 7.1580E-05 1.3995E-05 Aspergillus ibericus Engineered strain Fungi 0.002 0.001061181 9.2368E-05 7.3057E-05 Dichomitus squalens Engineered strain Fungi 0.002 0.018887472 8.0887E-05 4.1254E-05 Acinetobacter sp. TTH0-4 Engineered strain Bacteria Pseudomonadales 0.001333333 0.025523638 2.2311E-05 8.2612E-06 Rhizobium tropici Engineered strain Bacteria Rhizobiales 0.001333333 0.02079554 7.0081E-05 4.2000E-05 Methylocystis bryophila Engineered strain Bacteria Rhizobiales 0.001333333 0.006513543 3.5401E-05 2.2044E-05 Alteromonas naphthalenivorans Engineered strain Bacteria Alteromonadales 0.001 0.000660472 2.0747E-05 4.6463E-05 Saccharomyces cerevisiae Engineered strain Fungi 0.001 0.002980726 3.9901E-05 7.3043E-05 Bacillus phage Belinda Antibiotic Phage 0.002 0.016409765 6.8789E-07 6.0681E-08 Streptomyces sp. -

The Assembly and Functions of Microbial Communities on Complex Substrates
The assembly and functions of microbial communities on complex substrates by Xiaoqian Yu B.S./M.S., Molecular Biophysics and Biochemistry Yale University (2011) Submitted to the Biology Graduate Program in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy at the MASSACHUSETTS INSTITUTE OF TECHNOLOGY September 2019 2019 Massachusetts Institute of Technology. All rights reserved. Signature of Author: ____________________________________________________________________ Xiaoqian Yu Department of Biology Certified by: __________________________________________________________________________ Eric J. Alm Professor of Biological Engineering Professor of Civil and Environmental Engineering Thesis Advisor Accepted by: _________________________________________________________________________ Amy Keating Professor of Biology Co-Director, Biology Graduate Committee The assembly and functions of microbial communities on complex substrates by Xiaoqian Yu Submitted to the Department of Biology on August 5th, 2019 in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Biology Abstract Microbes form diverse and complex communities to influence the health and function of all ecosystems on earth. However, key ecological and evolutionary processes that allow microbial communities to form and maintain their diversity, and how this diversity further affects ecosystem function, are largely underexplored. This is especially true for natural microbial communities that harbor large numbers of species whose