Novel POC1A Mutation in Primordial Dwarfism Reveals New Insights For
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
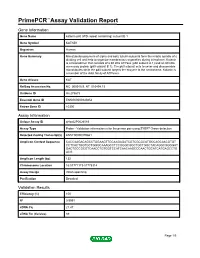
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name katanin p80 (WD repeat containing) subunit B 1 Gene Symbol KATNB1 Organism Human Gene Summary Microtubules polymers of alpha and beta tubulin subunits form the mitotic spindle of a dividing cell and help to organize membranous organelles during interphase. Katanin is a heterodimer that consists of a 60 kDa ATPase (p60 subunit A 1) and an 80 kDa accessory protein (p80 subunit B 1). The p60 subunit acts to sever and disassemble microtubules while the p80 subunit targets the enzyme to the centrosome. Katanin is a member of the AAA family of ATPases. Gene Aliases KAT RefSeq Accession No. NC_000016.9, NT_010498.15 UniGene ID Hs.275675 Ensembl Gene ID ENSG00000140854 Entrez Gene ID 10300 Assay Information Unique Assay ID qHsaCIP0026589 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000379661 Amplicon Context Sequence CACCAAGACAGCCTGGAAGTTGCAAGAGATCGTCGCGCATGCCAGCAACGTGT CCTCACTGGTGCTGGGCAAAGCCTCCGGGCGGCTGCTGGCTACAGGCGGGGAT GACTGCCGCGTCAACCTGTGGTCCATCAACAAGCCCAACTGCATCATGAGCCTG ACG Amplicon Length (bp) 132 Chromosome Location 16:57771173-57778314 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 100 R2 0.9991 cDNA Cq 21.47 cDNA Tm (Celsius) 89 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq 35.43 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report KATNB1, Human Amplification -

KIAA0556 Is a Novel Ciliary Basal Body Component Mutated in Joubert Syndrome Anna A
Sanders et al. Genome Biology (2015) 16:293 DOI 10.1186/s13059-015-0858-z RESEARCH Open Access KIAA0556 is a novel ciliary basal body component mutated in Joubert syndrome Anna A. W. M. Sanders1†, Erik de Vrieze2,3†, Anas M. Alazami4†, Fatema Alzahrani4, Erik B. Malarkey5, Nasrin Sorusch6, Lars Tebbe6, Stefanie Kuhns1, Teunis J. P. van Dam7, Amal Alhashem8, Brahim Tabarki8, Qianhao Lu9,10, Nils J. Lambacher1, Julie E. Kennedy1, Rachel V. Bowie1, Lisette Hetterschijt2,3, Sylvia van Beersum3,11, Jeroen van Reeuwijk3,11, Karsten Boldt12, Hannie Kremer2,3,11, Robert A. Kesterson13, Dorota Monies4, Mohamed Abouelhoda4, Ronald Roepman3,11, Martijn H. Huynen7, Marius Ueffing12, Rob B. Russell9,10, Uwe Wolfrum6, Bradley K. Yoder5, Erwin van Wijk2,3*, Fowzan S. Alkuraya4,14* and Oliver E. Blacque1* Abstract Background: Joubert syndrome (JBTS) and related disorders are defined by cerebellar malformation (molar tooth sign), together with neurological symptoms of variable expressivity. The ciliary basis of Joubert syndrome related disorders frequently extends the phenotype to tissues such as the eye, kidney, skeleton and craniofacial structures. Results: Using autozygome and exome analyses, we identified a null mutation in KIAA0556 in a multiplex consanguineous family with hallmark features of mild Joubert syndrome. Patient-derived fibroblasts displayed reduced ciliogenesis potential and abnormally elongated cilia. Investigation of disease pathophysiology revealed that Kiaa0556-/- null mice possess a Joubert syndrome-associated brain-restricted phenotype. Functional studies in Caenorhabditis elegans nematodes and cultured human cells support a conserved ciliary role for KIAA0556 linked to microtubule regulation. First, nematode KIAA0556 is expressed almost exclusively in ciliated cells, and the worm and human KIAA0556 proteins are enriched at the ciliary base. -

Katanin-P80 Gene Promoter Characterization and Regulation Via Elk1
Katanin-p80 Gene Promoter Characterization and Regulation via Elk1 Ece Selc¸uk., Koray Kırımtay., Derya Canbaz, Gu¨ her Is¸ık Cesur, Sirin Korulu, Arzu Karabay* Department of Molecular Biology and Genetics, Istanbul Technical University, Istanbul, Turkey Abstract Katanin is an ATPase family member protein that participates in microtubule severing. It has heterodimeric structure consisting of 60 kDa (katanin-p60) and 80 kDa (katanin-p80) subunits encoded by KATNA1 and KATNB1 genes, respectively. Katanin-p60 has the enzymatic activity for microtubule severing, whereas katanin-p80 consists of multiple domains with different functions such as targeting katanin-p60 to the centrosome, augmenting microtubule severing by katanin-p60, and even suppressing microtubule severing. Despite the various important functions of katanin-p80, its transcriptional regulation has not been studied yet. Elk1 transcription factor has been shown to interact with microtubules and regulate the transcription of another microtubule severing protein, spastin. In spite of katanin’s importance, and structural and functional similarities to spastin, there is no study on the transcriptional regulation of katanin yet. In this study, we aimed to characterize KATNB1 promoter and analyze the effects of Elk1 on katanin-p80 expression. We identified a 518- bp TATA-less promoter including a critical CpG island and GC boxes as an optimal promoter, and sequential deletion of CpG island and the GC elements gradually decreased the KATNB1 promoter activity. In addition, we showed Elk1 binding on the KATNB1 promoter by EMSA. We found that Elk1 activated KATNB1 promoter, and increased both mRNA and protein levels of katanin- p80 in SH-SY5Y cells. On the other hand, KCl treatment increasing SUMOylation decreased KATNB1 promoter activity. -

Supplemental Information
Supplemental information Dissection of the genomic structure of the miR-183/96/182 gene. Previously, we showed that the miR-183/96/182 cluster is an intergenic miRNA cluster, located in a ~60-kb interval between the genes encoding nuclear respiratory factor-1 (Nrf1) and ubiquitin-conjugating enzyme E2H (Ube2h) on mouse chr6qA3.3 (1). To start to uncover the genomic structure of the miR- 183/96/182 gene, we first studied genomic features around miR-183/96/182 in the UCSC genome browser (http://genome.UCSC.edu/), and identified two CpG islands 3.4-6.5 kb 5’ of pre-miR-183, the most 5’ miRNA of the cluster (Fig. 1A; Fig. S1 and Seq. S1). A cDNA clone, AK044220, located at 3.2-4.6 kb 5’ to pre-miR-183, encompasses the second CpG island (Fig. 1A; Fig. S1). We hypothesized that this cDNA clone was derived from 5’ exon(s) of the primary transcript of the miR-183/96/182 gene, as CpG islands are often associated with promoters (2). Supporting this hypothesis, multiple expressed sequences detected by gene-trap clones, including clone D016D06 (3, 4), were co-localized with the cDNA clone AK044220 (Fig. 1A; Fig. S1). Clone D016D06, deposited by the German GeneTrap Consortium (GGTC) (http://tikus.gsf.de) (3, 4), was derived from insertion of a retroviral construct, rFlpROSAβgeo in 129S2 ES cells (Fig. 1A and C). The rFlpROSAβgeo construct carries a promoterless reporter gene, the β−geo cassette - an in-frame fusion of the β-galactosidase and neomycin resistance (Neor) gene (5), with a splicing acceptor (SA) immediately upstream, and a polyA signal downstream of the β−geo cassette (Fig. -

Variation in Protein Coding Genes Identifies Information
bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 1 1 2 3 4 5 Variation in protein coding genes identifies information flow as a contributor to 6 animal complexity 7 8 Jack Dean, Daniela Lopes Cardoso and Colin Sharpe* 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 Institute of Biological and Biomedical Sciences 25 School of Biological Science 26 University of Portsmouth, 27 Portsmouth, UK 28 PO16 7YH 29 30 * Author for correspondence 31 [email protected] 32 33 Orcid numbers: 34 DLC: 0000-0003-2683-1745 35 CS: 0000-0002-5022-0840 36 37 38 39 40 41 42 43 44 45 46 47 48 49 Abstract bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 2 1 Across the metazoans there is a trend towards greater organismal complexity. How 2 complexity is generated, however, is uncertain. Since C.elegans and humans have 3 approximately the same number of genes, the explanation will depend on how genes are 4 used, rather than their absolute number. -

Retroviral Vector Integration Deregulates Gene Expression but Has No Consequence on the Biology and Function of Transplanted T Cells
Retroviral vector integration deregulates gene expression but has no consequence on the biology and function of transplanted T cells Alessandra Recchia*†, Chiara Bonini*‡, Zulma Magnani*, Fabrizia Urbinati†, Daniela Sartori*§, Sara Muraro*, Enrico Tagliafico†, Attilio Bondanza*¶, Maria Teresa Lupo Stanghellini‡, Massimo Bernardi‡, Alessandra Pescarollo‡, Fabio Ciceri‡, Claudio Bordignon§¶, and Fulvio Mavilio†ʈ *Cancer Immunotherapy and Gene Therapy Program, and ‡Bone Marrow Transplantation Unit, Istituto Scientifico H. San Raffaele, Via Olgettina 58, 20132 Milan, Italy; †Department of Biomedical Sciences, University of Modena and Reggio Emilia, Via Campi 287, 41100 Modena, Italy; §MolMed S.p.A., Via Olgettina 58, 20132 Milan, Italy; and ¶San Raffaele ‘‘Vita e Salute’’ University, Via Olgettina 58, 20132 Milan, Italy Edited by Malcolm A. Martin, National Institutes of Health, Bethesda, MD, and approved December 6, 2005 (received for review September 1, 2005) The use of retroviral vectors in gene therapy has raised safety is currently profiled to provide a graft-versus-leukemia effect to concerns for the genotoxic risk associated with their uncontrolled patients in relapse after HLA-identical HSC transplantation (14), insertion into the human genome. We have analyzed the conse- or to promote immune reconstitution and prevent relapse in quences of retroviral transduction in T cells from leukemic patients patients undergoing HLA-haploidentical HSC transplantation. In treated with allogeneic stem cell transplantation and donor lym- 46 patients treated since 1994 in both contexts, we were able to phocytes genetically modified with a suicide gene (HSV-TK). Ret- control GvHD in 100% of the cases, while preserving antiviral and roviral vectors integrate preferentially within or near transcribed antitumor activity (14–16). -

Host Cell Factors Necessary for Influenza a Infection: Meta-Analysis of Genome Wide Studies
Host Cell Factors Necessary for Influenza A Infection: Meta-Analysis of Genome Wide Studies Juliana S. Capitanio and Richard W. Wozniak Department of Cell Biology, Faculty of Medicine and Dentistry, University of Alberta Abstract: The Influenza A virus belongs to the Orthomyxoviridae family. Influenza virus infection occurs yearly in all countries of the world. It usually kills between 250,000 and 500,000 people and causes severe illness in millions more. Over the last century alone we have seen 3 global influenza pandemics. The great human and financial cost of this disease has made it the second most studied virus today, behind HIV. Recently, several genome-wide RNA interference studies have focused on identifying host molecules that participate in Influen- za infection. We used nine of these studies for this meta-analysis. Even though the overlap among genes identified in multiple screens was small, network analysis indicates that similar protein complexes and biological functions of the host were present. As a result, several host gene complexes important for the Influenza virus life cycle were identified. The biological function and the relevance of each identified protein complex in the Influenza virus life cycle is further detailed in this paper. Background and PA bound to the viral genome via nucleoprotein (NP). The viral core is enveloped by a lipid membrane derived from Influenza virus the host cell. The viral protein M1 underlies the membrane and anchors NEP/NS2. Hemagglutinin (HA), neuraminidase Viruses are the simplest life form on earth. They parasite host (NA), and M2 proteins are inserted into the envelope, facing organisms and subvert the host cellular machinery for differ- the viral exterior. -

A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family
Rockefeller University Digital Commons @ RU Student Theses and Dissertations 2018 A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family Linda Molla Follow this and additional works at: https://digitalcommons.rockefeller.edu/ student_theses_and_dissertations Part of the Life Sciences Commons A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY A Thesis Presented to the Faculty of The Rockefeller University in Partial Fulfillment of the Requirements for the degree of Doctor of Philosophy by Linda Molla June 2018 © Copyright by Linda Molla 2018 A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY Linda Molla, Ph.D. The Rockefeller University 2018 APOBEC2 is a member of the AID/APOBEC cytidine deaminase family of proteins. Unlike most of AID/APOBEC, however, APOBEC2’s function remains elusive. Previous research has implicated APOBEC2 in diverse organisms and cellular processes such as muscle biology (in Mus musculus), regeneration (in Danio rerio), and development (in Xenopus laevis). APOBEC2 has also been implicated in cancer. However the enzymatic activity, substrate or physiological target(s) of APOBEC2 are unknown. For this thesis, I have combined Next Generation Sequencing (NGS) techniques with state-of-the-art molecular biology to determine the physiological targets of APOBEC2. Using a cell culture muscle differentiation system, and RNA sequencing (RNA-Seq) by polyA capture, I demonstrated that unlike the AID/APOBEC family member APOBEC1, APOBEC2 is not an RNA editor. Using the same system combined with enhanced Reduced Representation Bisulfite Sequencing (eRRBS) analyses I showed that, unlike the AID/APOBEC family member AID, APOBEC2 does not act as a 5-methyl-C deaminase. -

Regulation of Human Cerebral Cortical Development by EXOC7
ARTICLE Regulation of human cerebral cortical development by EXOC7 and EXOC8, components of the exocyst complex, and roles in neural progenitor cell proliferation and survival Michael E. Coulter, MD, PhD1,2, Damir Musaev, BSc3, Ellen M. DeGennaro, BA1,4, Xiaochang Zhang, PhD1,5, Katrin Henke, PhD6, Kiely N. James, PhD3, Richard S. Smith, PhD1, R. Sean Hill, PhD1, Jennifer N. Partlow, MS1, Muna Al-Saffar, MBChB, MSc1,7, A. Stacy Kamumbu, BA1, Nicole Hatem, BA1, A. James Barkovich, MD8, Jacqueline Aziza, MD9, Nicolas Chassaing, MD, PhD10,11, Maha S. Zaki, MD, PhD12, Tipu Sultan, MD13, Lydie Burglen, MD, PhD14,15, Anna Rajab, MD, PhD16, Lihadh Al-Gazali, MBChB, MSc7, Ganeshwaran H. Mochida, MD, MMSc1,17, Matthew P. Harris, PhD6, Joseph G. Gleeson, MD3 and Christopher A. Walsh, MD, PhD 1 Purpose: The exocyst complex is a conserved protein complex that (LOF) variants in a recessively inherited disorder characterized by mediates fusion of intracellular vesicles to the plasma membrane brain atrophy, seizures, and developmental delay, and in severe and is implicated in processes including cell polarity, cell migration, cases, microcephaly and infantile death. In EXOC8, we found a ciliogenesis, cytokinesis, autophagy, and fusion of secretory vesicles. homozygous truncating variant in a family with a similar clinical The essential role of these genes in human genetic disorders, disorder. We modeled exoc7 deficiency in zebrafish and found the however, is unknown. absence of exoc7 causes microcephaly. Methods: We performed homozygosity mapping and exome Conclusion: Our results highlight the essential role of the exocyst sequencing of consanguineous families with recessively inherited pathway in normal cortical development and how its perturbation brain development disorders. -

SPEF2 Functions in Microtubule-Mediated Transport in Elongating Spermatids to Ensure Proper Male Germ Cell Differentiation Mari S
© 2017. Published by The Company of Biologists Ltd | Development (2017) 144, 2683-2693 doi:10.1242/dev.152108 RESEARCH ARTICLE SPEF2 functions in microtubule-mediated transport in elongating spermatids to ensure proper male germ cell differentiation Mari S. Lehti1,2,*, Fu-Ping Zhang2,3,*, Noora Kotaja2,* and Anu Sironen1,‡,§ ABSTRACT 2006, 2002). Mutations in the Spef2 gene (an amino acid substitution Sperm differentiation requires specific protein transport for correct within exon 3 and a nonsense mutation within exon 28) in the big giant sperm tail formation and head shaping. A transient microtubular head (bgh) mouse model caused a primary ciliary dyskinesia (PCD)- structure, the manchette, appears around the differentiating like phenotype, including hydrocephalus, sinusitis and male infertility spermatid head and serves as a platform for protein transport to the (Sironen et al., 2011). Detailed analysis of spermatogenesis in both pig growing tail. Sperm flagellar 2 (SPEF2) is known to be essential for and mouse models revealed axonemal abnormalities, including defects sperm tail development. In this study we investigated the function of in central pair (CP) structure and the complete disorganization of the SPEF2 during spermatogenesis using a male germ cell-specific sperm tail (Sironen et al., 2011). A role of SPEF2 in protein transport Spef2 knockout mouse model. In addition to defects in sperm tail has been postulated owing to its known interaction and colocalization development, we observed a duplication of the basal body and failure with intraflagellar transport 20 (IFT20). During spermatogenesis, in manchette migration resulting in an abnormal head shape. We IFT20 and SPEF2 colocalize in the Golgi complex of late identified cytoplasmic dynein 1 and GOLGA3 as novel interaction spermatocytes and round spermatids and in the manchette and basal partners for SPEF2. -

Supplementary Table 1 Double Treatment Vs Single Treatment
Supplementary table 1 Double treatment vs single treatment Probe ID Symbol Gene name P value Fold change TC0500007292.hg.1 NIM1K NIM1 serine/threonine protein kinase 1.05E-04 5.02 HTA2-neg-47424007_st NA NA 3.44E-03 4.11 HTA2-pos-3475282_st NA NA 3.30E-03 3.24 TC0X00007013.hg.1 MPC1L mitochondrial pyruvate carrier 1-like 5.22E-03 3.21 TC0200010447.hg.1 CASP8 caspase 8, apoptosis-related cysteine peptidase 3.54E-03 2.46 TC0400008390.hg.1 LRIT3 leucine-rich repeat, immunoglobulin-like and transmembrane domains 3 1.86E-03 2.41 TC1700011905.hg.1 DNAH17 dynein, axonemal, heavy chain 17 1.81E-04 2.40 TC0600012064.hg.1 GCM1 glial cells missing homolog 1 (Drosophila) 2.81E-03 2.39 TC0100015789.hg.1 POGZ Transcript Identified by AceView, Entrez Gene ID(s) 23126 3.64E-04 2.38 TC1300010039.hg.1 NEK5 NIMA-related kinase 5 3.39E-03 2.36 TC0900008222.hg.1 STX17 syntaxin 17 1.08E-03 2.29 TC1700012355.hg.1 KRBA2 KRAB-A domain containing 2 5.98E-03 2.28 HTA2-neg-47424044_st NA NA 5.94E-03 2.24 HTA2-neg-47424360_st NA NA 2.12E-03 2.22 TC0800010802.hg.1 C8orf89 chromosome 8 open reading frame 89 6.51E-04 2.20 TC1500010745.hg.1 POLR2M polymerase (RNA) II (DNA directed) polypeptide M 5.19E-03 2.20 TC1500007409.hg.1 GCNT3 glucosaminyl (N-acetyl) transferase 3, mucin type 6.48E-03 2.17 TC2200007132.hg.1 RFPL3 ret finger protein-like 3 5.91E-05 2.17 HTA2-neg-47424024_st NA NA 2.45E-03 2.16 TC0200010474.hg.1 KIAA2012 KIAA2012 5.20E-03 2.16 TC1100007216.hg.1 PRRG4 proline rich Gla (G-carboxyglutamic acid) 4 (transmembrane) 7.43E-03 2.15 TC0400012977.hg.1 SH3D19 -

Program Book
The Genetics Society of America Conferences 15th International Xenopus Conference August 24-28, 2014 • Pacific Grove, CA PROGRAM GUIDE LEGEND Information/Guest Check-In Disabled Parking E EV Charging Station V Beverage Vending Machine N S Ice Machine Julia Morgan Historic Building W Roadway Pedestrian Pathway Outdoor Group Activity Area Program and Abstracts Meeting Organizers Carole LaBonne, Northwestern University John Wallingford, University of Texas at Austin Organizing Committee: Julie Baker, Stanford Univ Chris Field, Harvard Medical School Carmen Domingo, San Francisco State Univ Anna Philpott, Univ of Cambridge 9650 Rockville Pike, Bethesda, Maryland 20814-3998 Telephone: (301) 634-7300 • Fax: (301) 634-7079 E-mail: [email protected] • Web site: genetics-gsa.org Thank You to the Following Companies for their Generous Support Platinum Sponsor Gold Sponsors Additional Support Provided by: Carl Zeiss Microscopy, LLC Monterey Convention & Gene Tools, LLC Visitors Bureau Leica Microsystems Xenopus Express 2 Table of Contents General Information ........................................................................................................................... 5 Schedule of Events ............................................................................................................................. 6 Exhibitors ........................................................................................................................................... 8 Opening Session and Plenary/Platform Sessions ............................................................................