Drosophila SWR1 and Nua4 Complexes Are Defined by DOMINO
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Download Download
Supplementary Figure S1. Results of flow cytometry analysis, performed to estimate CD34 positivity, after immunomagnetic separation in two different experiments. As monoclonal antibody for labeling the sample, the fluorescein isothiocyanate (FITC)- conjugated mouse anti-human CD34 MoAb (Mylteni) was used. Briefly, cell samples were incubated in the presence of the indicated MoAbs, at the proper dilution, in PBS containing 5% FCS and 1% Fc receptor (FcR) blocking reagent (Miltenyi) for 30 min at 4 C. Cells were then washed twice, resuspended with PBS and analyzed by a Coulter Epics XL (Coulter Electronics Inc., Hialeah, FL, USA) flow cytometer. only use Non-commercial 1 Supplementary Table S1. Complete list of the datasets used in this study and their sources. GEO Total samples Geo selected GEO accession of used Platform Reference series in series samples samples GSM142565 GSM142566 GSM142567 GSM142568 GSE6146 HG-U133A 14 8 - GSM142569 GSM142571 GSM142572 GSM142574 GSM51391 GSM51392 GSE2666 HG-U133A 36 4 1 GSM51393 GSM51394 only GSM321583 GSE12803 HG-U133A 20 3 GSM321584 2 GSM321585 use Promyelocytes_1 Promyelocytes_2 Promyelocytes_3 Promyelocytes_4 HG-U133A 8 8 3 GSE64282 Promyelocytes_5 Promyelocytes_6 Promyelocytes_7 Promyelocytes_8 Non-commercial 2 Supplementary Table S2. Chromosomal regions up-regulated in CD34+ samples as identified by the LAP procedure with the two-class statistics coded in the PREDA R package and an FDR threshold of 0.5. Functional enrichment analysis has been performed using DAVID (http://david.abcc.ncifcrf.gov/) -

ING3 Is Essential for Asymmetric Cell Division During Mouse Oocyte Maturation
ING3 Is Essential for Asymmetric Cell Division during Mouse Oocyte Maturation Shinnosuke Suzuki1, Yusuke Nozawa1, Satoshi Tsukamoto2, Takehito Kaneko3, Hiroshi Imai1, Naojiro Minami1* 1 Laboratory of Reproductive Biology, Graduate School of Agriculture, Kyoto University, Kyoto, Japan, 2 Laboratory Animal and Genome Sciences Section, National Institute of Radiological Sciences, Chiba, Japan, 3 Institute of Laboratory Animals, Graduate School of Medicine, Kyoto University, Kyoto, Japan Abstract ING3 (inhibitor of growth family, member 3) is a subunit of the nucleosome acetyltransferase of histone 4 (NuA4) complex, which activates gene expression. ING3, which contains a plant homeodomain (PHD) motif that can bind to trimethylated lysine 4 on histone H3 (H3K4me3), is ubiquitously expressed in mammalian tissues and governs transcriptional regulation, cell cycle control, and apoptosis via p53-mediated transcription or the Fas/caspase-8 pathway. Thus, ING3 plays a number of important roles in various somatic cells. However, the role(s) of ING3 in germ cells remains unknown. Here, we show that loss of ING3 function led to the failure of asymmetric cell division and cortical reorganization in the mouse oocyte. Immunostaining showed that in fully grown germinal vesicle (GV) oocytes, ING3 localized predominantly in the GV. After germinal vesicle breakdown (GVBD), ING3 homogeneously localized in the cytoplasm. In oocytes where Ing3 was targeted by siRNA microinjection, we observed symmetric cell division during mouse oocyte maturation. In those oocytes, oocyte polarization was not established due to the failure to form an actin cap or a cortical granule-free domain (CGFD), the lack of which inhibited spindle migration. These features were among the main causes of abnormal symmetric cell division. -

Supporting Information
Supporting Information Pouryahya et al. SI Text Table S1 presents genes with the highest absolute value of Ricci curvature. We expect these genes to have significant contribution to the network’s robustness. Notably, the top two genes are TP53 (tumor protein 53) and YWHAG gene. TP53, also known as p53, it is a well known tumor suppressor gene known as the "guardian of the genome“ given the essential role it plays in genetic stability and prevention of cancer formation (1, 2). Mutations in this gene play a role in all stages of malignant transformation including tumor initiation, promotion, aggressiveness, and metastasis (3). Mutations of this gene are present in more than 50% of human cancers, making it the most common genetic event in human cancer (4, 5). Namely, p53 mutations play roles in leukemia, breast cancer, CNS cancers, and lung cancers, among many others (6–9). The YWHAG gene encodes the 14-3-3 protein gamma, a member of the 14-3-3 family proteins which are involved in many biological processes including signal transduction regulation, cell cycle pro- gression, apoptosis, cell adhesion and migration (10, 11). Notably, increased expression of 14-3-3 family proteins, including protein gamma, have been observed in a number of human cancers including lung and colorectal cancers, among others, suggesting a potential role as tumor oncogenes (12, 13). Furthermore, there is evidence that loss Fig. S1. The histogram of scalar Ricci curvature of 8240 genes. Most of the genes have negative scalar Ricci curvature (75%). TP53 and YWHAG have notably low of p53 function may result in upregulation of 14-3-3γ in lung cancer Ricci curvatures. -

(P -Value<0.05, Fold Change≥1.4), 4 Vs. 0 Gy Irradiation
Table S1: Significant differentially expressed genes (P -Value<0.05, Fold Change≥1.4), 4 vs. 0 Gy irradiation Genbank Fold Change P -Value Gene Symbol Description Accession Q9F8M7_CARHY (Q9F8M7) DTDP-glucose 4,6-dehydratase (Fragment), partial (9%) 6.70 0.017399678 THC2699065 [THC2719287] 5.53 0.003379195 BC013657 BC013657 Homo sapiens cDNA clone IMAGE:4152983, partial cds. [BC013657] 5.10 0.024641735 THC2750781 Ciliary dynein heavy chain 5 (Axonemal beta dynein heavy chain 5) (HL1). 4.07 0.04353262 DNAH5 [Source:Uniprot/SWISSPROT;Acc:Q8TE73] [ENST00000382416] 3.81 0.002855909 NM_145263 SPATA18 Homo sapiens spermatogenesis associated 18 homolog (rat) (SPATA18), mRNA [NM_145263] AA418814 zw01a02.s1 Soares_NhHMPu_S1 Homo sapiens cDNA clone IMAGE:767978 3', 3.69 0.03203913 AA418814 AA418814 mRNA sequence [AA418814] AL356953 leucine-rich repeat-containing G protein-coupled receptor 6 {Homo sapiens} (exp=0; 3.63 0.0277936 THC2705989 wgp=1; cg=0), partial (4%) [THC2752981] AA484677 ne64a07.s1 NCI_CGAP_Alv1 Homo sapiens cDNA clone IMAGE:909012, mRNA 3.63 0.027098073 AA484677 AA484677 sequence [AA484677] oe06h09.s1 NCI_CGAP_Ov2 Homo sapiens cDNA clone IMAGE:1385153, mRNA sequence 3.48 0.04468495 AA837799 AA837799 [AA837799] Homo sapiens hypothetical protein LOC340109, mRNA (cDNA clone IMAGE:5578073), partial 3.27 0.031178378 BC039509 LOC643401 cds. [BC039509] Homo sapiens Fas (TNF receptor superfamily, member 6) (FAS), transcript variant 1, mRNA 3.24 0.022156298 NM_000043 FAS [NM_000043] 3.20 0.021043295 A_32_P125056 BF803942 CM2-CI0135-021100-477-g08 CI0135 Homo sapiens cDNA, mRNA sequence 3.04 0.043389246 BF803942 BF803942 [BF803942] 3.03 0.002430239 NM_015920 RPS27L Homo sapiens ribosomal protein S27-like (RPS27L), mRNA [NM_015920] Homo sapiens tumor necrosis factor receptor superfamily, member 10c, decoy without an 2.98 0.021202829 NM_003841 TNFRSF10C intracellular domain (TNFRSF10C), mRNA [NM_003841] 2.97 0.03243901 AB002384 C6orf32 Homo sapiens mRNA for KIAA0386 gene, partial cds. -

Overexpression of ING3 Is Associated with Attenuation of Migration and Invasion in Breast Cancer
EXPERIMENTAL AND THERAPEUTIC MEDICINE 22: 699, 2021 Overexpression of ING3 is associated with attenuation of migration and invasion in breast cancer HUIMENG LI1*, HENGYU ZHANG1*, XIN TAN1*, DEQUAN LIU1, RONG GUO1, MAOHUA WANG1, YIYIN TANG1, KAI ZHENG1, WENLIN CHEN1, HONGWAN LI1, MINGJIAN TAN1, KE WANG1, RUI LIU2 and SHICONG TANG1 1Department of Breast Surgery, The Third Affiliated Hospital of Kunming Medical University, Yunnan Cancer Hospital, Kunming, Yunnan 650118; 2Department of Clinical Laboratory, The Second Affiliated Hospital of Kunming Medical University, Kunming, Yunnan 650101, P.R. China Received May 9, 2020; Accepted March 24, 2021 DOI: 10.3892/etm.2021.10131 Abstract. Inhibitor of growth 3 (ING3) has been identified Notably, statistically significant differences were observed as a potential cancer drug target, but little is known about its in long‑term survival between patients with luminal A role in breast cancer. Thus, the present study aimed to inves‑ (P=0.04) and HER2‑enriched (P=0.008) breast cancer, with tigate ING3 expression in breast cancer, its clinical value, and high and low expression levels of ING3. The results of the how ING3 influences the migration and invasion of breast Transwell migration and invasion assays demonstrated that cancer cells. The Cancer Genome Atlas and UALCAN data‑ overexpression of ING3 significantly inhibited the migra‑ bases were used to analyze ING3 expression in cancer tissues tory and invasive abilities of MCF7 (P<0.05) and HCC1937 and normal tissues. Survival analysis was performed using the (P<0.05) cells. The results of the wound healing assay UALCAN, UCSC Xena and KM‑plot databases. -

Gene Section Review
Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL INIST-CNRS Gene Section Review ING3 (inhibitor of growth family, member 3) Audrey Mouche, Katherine Yaacoub, Thierry Guillaudeux, Rémy Pedeux INSERM U917, Microenvironnement et Cancer, Rennes, France, Universite de Rennes 1, Rennes, France (AM, KY, RP, TG); Etablissement Francais du Sang, Rennes, France (RP); UMS Biosit 3480 CNRS/018 INSERM (TG) Published in Atlas Database: November 2014 Online updated version : http://AtlasGeneticsOncology.org/Genes/ING3ID40977ch7q31.html Printable original version : http://documents.irevues.inist.fr/bitstream/handle/2042/62489/11-2014-ING3ID40977ch7q31.pdf DOI: 10.4267/2042/62489 This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2015 Atlas of Genetics and Cytogenetics in Oncology and Haematology Abstract Review on ING3, with data on DNA/RNA, on the protein encoded and where the gene is implicated. Identity Other names: Eaf4, ING2, MEAF4, p47ING3 HGNC (Hugo): ING3 Location: 7q31.31 DNA/RNA Description In 1996, Karl Riabowol's group identified a new Tumor Suppressor Gene (TSG) by using subtractive hybridization between cDNAs from normal mammary epithelial cells and mammary epithelial cells from tumor. This experiment was followed by an in vivo screen for tumourigenesis. Using this method, the authors identified a new candidate TSG that they named ING1 for INhibitor of Growth 1 (Garkavtsev et al., 1996). Few years later, ING2, ING3, ING4 and ING5 were identified by homology search. ING3 was identified through bioinformatic analyses in order to find human EST clone showing Figure 1. Chromosomal localization of the ING3 gene in a high homology with the p33ING1b and p33ING2 Homo sapiens. -

ING3 (NM 019071) Human Tagged ORF Clone Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC218424 ING3 (NM_019071) Human Tagged ORF Clone Product data: Product Type: Expression Plasmids Product Name: ING3 (NM_019071) Human Tagged ORF Clone Tag: Myc-DDK Symbol: ING3 Synonyms: Eaf4; ING2; MEAF4; p47ING3 Vector: pCMV6-Entry (PS100001) E. coli Selection: Kanamycin (25 ug/mL) Cell Selection: Neomycin ORF Nucleotide >RC218424 representing NM_019071 Sequence: Red=Cloning site Blue=ORF Green=Tags(s) TTTTGTAATACGACTCACTATAGGGCGGCCGGGAATTCGTCGACTGGATCCGGTACCGAGGAGATCTGCC GCCGCGATCGCC ATGTTGTACCTAGAAGACTATCTGGAAATGATTGAGCAGCTTCCTATGGATCTGCGGGACCGCTTCACGG AAATGCGCGAGATGGACCTGCAGGTGCAGAATGCAATGGATCAACTAGAACAAAGAGTCAGTGAATTCTT TATGAATGCAAAGAAAAATAAACCTGAGTGGAGGGAAGAGCAAATGGCATCCATCAAAAAAGACTACTAT AAAGCTTTGGAAGATGCAGATGAGAAGGTTCAGTTGGCAAACCAGATATATGACTTGGTAGATCGACACT TGAGAAAGCTGGATCAGGAACTGGCTAAGTTTAAAATGGAGCTGGAAGCTGATAATGCTGGAATTACAGA AATATTAGAGAGGCGATCTTTGGAATTAGACACTCCTTCACAGCCAGTGAACAATCACCATGCTCATTCA CATACTCCAGTGGAAAAAAGGAAATATAATCCAACTTCTCACCATACGACAACAGATCATATTCCTGAAA AGAAATTTAAATCTGAAGCTCTTCTATCCACCCTTACGTCAGATGCCTCTAAGGAAAATACACTAGGTTG TCGAAATAATAATTCCACAGCCTCTTCTAACAATGCCTACAATGTGAATTCCTCCCAACCTCTGGGATCC TATAACATTGGCTCGTTATCTTCAGGAACTGGTGCAGGGGCAATTACCATGGCAGCTGCTCAAGCAGTTC AGGCTACAGCTCAGATGAAGGAGGGACGAAGAACATCAAGTTTAAAAGCCAGTTATGAAGCATTTAAGAA TAATGACTTTCAGTTGGGAAAAGAATTTTCAATGGCCAGGGAAACAGTTGGCTATTCATCATCTTCGGCA CTTATGACAACATTAACACAGAATGCCAGTTCATCAGCAGCCGACTCACGGAGTGGTCGAAAGAGCAAAA -
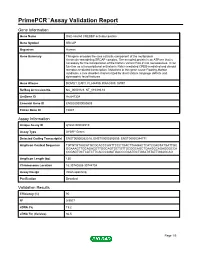
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name Snf2-related CREBBP activator protein Gene Symbol SRCAP Organism Human Gene Summary This gene encodes the core catalytic component of the multiprotein chromatin-remodeling SRCAP complex. The encoded protein is an ATPase that is necessary for the incorporation of the histone variant H2A.Z into nucleosomes. It can function as a transcriptional activator in Notch-mediated CREB-mediated and steroid receptor-mediated transcription. Mutations in this gene cause Floating-Harbor syndrome a rare disorder characterized by short stature language deficits and dysmorphic facial features. Gene Aliases DOMO1, EAF1, FLJ44499, KIAA0309, SWR1 RefSeq Accession No. NC_000016.9, NT_010393.16 UniGene ID Hs.647334 Ensembl Gene ID ENSG00000080603 Entrez Gene ID 10847 Assay Information Unique Assay ID qHsaCID0006510 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000262518, ENST00000395059, ENST00000344771 Amplicon Context Sequence TGTGTGTAACATGCGCACCCAGTTCCCTGACTTAAGACTCATCCAGTATGATTGC GGAAAGTTGCAGACGTTGGCAGTGCTGTTGCGGCAGCTCAAGGCAGAGGGCCA CCGAGTGCTCATCTTCACCCAGATGACCCGAATGCTGGATGTATTGGAGCAG Amplicon Length (bp) 130 Chromosome Location 16:30740838-30744704 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 90 R2 0.9977 cDNA Cq 19.2 cDNA Tm (Celsius) 86.5 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report SRCAP, Human -

Targeted Disruption of the Mouse Ing1 Locus Results in Reduced Body Size, Hypersensitivity to Radiation and Elevated Incidence of Lymphomas
Oncogene (2006) 25, 857–866 & 2006 Nature Publishing Group All rights reserved 0950-9232/06 $30.00 www.nature.com/onc ORIGINAL ARTICLE Targeted disruption of the mouse ing1 locus results in reduced body size, hypersensitivity to radiation and elevated incidence of lymphomas JV Kichina1,2,6, M Zeremski2,6,7, L Aris2,6, KV Gurova1,3, E Walker4, RFranks 2, AY Nikitin5, H Kiyokawa2 and AV Gudkov1,3 1Department of Molecular Genetics, Lerner Research Institute, Cleveland, OH, USA; 2Department of Biochemistry and Molecular Genetics, University of Illinois at Chicago, Chicago, IL, USA; 3Cleveland BioLabs, Inc., Cleveland, OH, USA; 4Department of Quantitative Health Sciences, The Cleveland Clinic Foundation, Cleveland, OH, USA and 5Department of Biomedical Sciences, Cornell University, Ithaca, NY, USA Ing1 belongs to the family of evolutionary conserved genes neoplastic transformation of mouse mammary epithelial encoding nuclear PHD finger-containing proteins impli- cells: the GSE that encoded an antisense RNA against a cated in a variety of processes, including tumorigenesis, previously unknown gene promoted transformation of replicative senescence, excision repair and response to fibroblasts and epithelia cells (Garkavtsev et al., 1996). genotoxic stress. We have generated mice deficient in all Since overexpression of cDNA for the identified gene the isoforms of Ing1 by targeted disruption of the exon resulted in growth repression of human fibroblasts, it that is common for all ing1 transcripts. Embryonic was named Ing1 (abbreviation from ‘INhibitor of fibroblasts from ing1-knockout mice were similar to the Growth’; Garkavtsev et al. (1996)). Decreased expres- wild-type cells in their growth characteristics, replicative sion of ING1 was found in several breast carcinomas lifespan in culture, p53 induction and sensitivity to various and the Ing1 gene was found rearranged in one cytotoxic treatments with minor alterations in cell cycle neuroblastoma cell line pointing at its putative tumor- distribution in response to genotoxic stress. -

The Role of the ING3 Epigenetic Regulator in Prostate Cancer
University of Calgary PRISM: University of Calgary's Digital Repository Graduate Studies The Vault: Electronic Theses and Dissertations 2017 The Role of the ING3 Epigenetic Regulator in Prostate Cancer Nabbi, Arash Nabbi, A. (2017). The Role of the ING3 Epigenetic Regulator in Prostate Cancer (Unpublished doctoral thesis). University of Calgary, Calgary, AB. doi:10.11575/PRISM/28360 http://hdl.handle.net/11023/3584 doctoral thesis University of Calgary graduate students retain copyright ownership and moral rights for their thesis. You may use this material in any way that is permitted by the Copyright Act or through licensing that has been assigned to the document. For uses that are not allowable under copyright legislation or licensing, you are required to seek permission. Downloaded from PRISM: https://prism.ucalgary.ca UNIVERSITY OF CALGARY The Role of the ING3 Epigenetic Regulator in Prostate Cancer by Arash Nabbi A THESIS SUBMITTED TO THE FACULTY OF GRADUATE STUDIES IN PARTIAL FULFILMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY GRADUATE PROGRAM IN MEDICAL SCIENCE CALGARY, ALBERTA JANUARY, 2017 © Arash Nabbi 2017 Abstract INhibitor of growth (ING) proteins are epigenetic regulators and stoichiometric members of histone acetyltransferase (KAT) or histone deacetylase (KDAC) complexes. By reading the histone mark H3K4me3, they direct their complexes to chromatin to alter gene expression. This thesis focuses on the role of ING3 in prostate cancer biology. Since rigorous characterization of antibodies is a prerequisite to acquire reliable results, we began by characterizing a new mouse monoclonal antibody against ING3. We profiled the expression of ING3 protein in normal human tissues and found that it is highly expressed in bone marrow, suggesting high expression in hematopoietic cell precursors. -

Computational Analysis of Protein Function Within Complete Genomes
Computational Analysis of Protein Function within Complete Genomes Anton James Enright Wolfson College A dissertation submitted to the University of Cambridge for the degree of Doctor of Philosophy European Molecular Biology Laboratory, European Bioinformatics Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, CB10 1SD, United Kingdom. Email: [email protected] March 7, 2002 To My Parents and Kerstin This thesis is the result of my own work and includes nothing which is the outcome of work done in collaboration except where specifically indicated in the text. This thesis does not exceed the specified length limit of 300 pages as de- fined by the Biology Degree Committee. This thesis has been typeset in 12pt font using LATEX2ε accordingtothe specifications defined by the Board of Graduate Studies and the Biology Degree Committee. ii Computational Analysis of Protein Function within Complete Genomes Summary Anton James Enright March 7, 2002 Wolfson College Since the advent of complete genome sequencing, vast amounts of nucleotide and amino acid sequence data have been produced. These data need to be effectively analysed and verified so that they may be used for biologi- cal discovery. A significant proportion of predicted protein sequences from these complete genomes have poorly characterised or unknown functional annotations. This thesis describes a number of approaches which detail the computational analysis of amino acid sequences for the prediction and analy- sis of protein function within complete genomes. The first chapter is a short introduction to computational genome analysis while the second and third chapters describe how groups of related protein sequences (termed protein families) may be characterised using sequence clustering algorithms. -

Dynamics of Transcription Elongation Are Finely Tuned by Dozens of Regulatory Factors
bioRxiv preprint doi: https://doi.org/10.1101/2021.08.15.456358; this version posted August 15, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Dynamics of transcription elongation are finely tuned by dozens of regulatory factors Mary Couvillion1*, Kevin M. Harlen1*, Kate C. Lachance1*, Kristine L. Trotta1, Erin Smith1, Christian Brion1, Brendan M. Smalec1, L. Stirling Churchman1 1Blavatnik Institute, Department of Genetics, Harvard Medical School, Boston, Massachusetts, USA 02115 *these authors contributed equally 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.08.15.456358; this version posted August 15, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. ABSTRACT Understanding the complex network and dynamics that regulate transcription elongation requires the quantitative analysis of RNA polymerase II (Pol II) activity in a wide variety of regulatory environments. We performed native elongating transcript sequencing (NET-seq) in 41 strains of S. cerevisiae lacking known elongation regulators, including RNA processing factors, transcription elongation factors, chromatin modifiers, and remodelers. We found that the opposing effects of these factors balance transcription elongation dynamics. Different sets of factors tightly regulate Pol II progression across gene bodies so that Pol II density peaks at key points of RNA processing.