Qt47b6d6d2.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Spatial Protein Interaction Networks of the Intrinsically Disordered Transcription Factor C(%3$
Spatial protein interaction networks of the intrinsically disordered transcription factor C(%3$ Dissertation zur Erlangung des akademischen Grades Doctor rerum naturalium (Dr. rer. nat.) im Fach Biologie/Molekularbiologie eingereicht an der Lebenswissenschaftlichen Fakultät der Humboldt-Universität zu Berlin Von Evelyn Ramberger, M.Sc. Präsidentin der Humboldt-Universität zu Berlin Prof. Dr.-Ing.Dr. Sabine Kunst Dekan der Lebenswissenschaftlichen Fakultät der Humboldt-Universität zu Berlin Prof. Dr. Bernhard Grimm Gutachter: 1. Prof. Dr. Achim Leutz 2. Prof. Dr. Matthias Selbach 3. Prof. Dr. Gunnar Dittmar Tag der mündlichen Prüfung: 12.8.2020 For T. Table of Contents Selbstständigkeitserklärung ....................................................................................1 List of Figures ............................................................................................................2 List of Tables ..............................................................................................................3 Abbreviations .............................................................................................................4 Zusammenfassung ....................................................................................................6 Summary ....................................................................................................................7 1. Introduction ............................................................................................................8 1.1. Disordered proteins -
Identification of 42 Genes Linked to Stage II Colorectal Cancer Metastatic Relapse
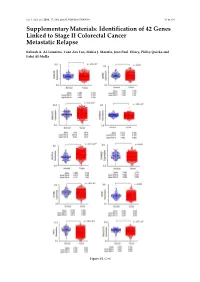
Int. J. Mol. Sci. 2016, 17, 598; doi:10.3390/ijms17040598 S1 of S16 Supplementary Materials: Identification of 42 Genes Linked to Stage II Colorectal Cancer Metastatic Relapse Rabeah A. Al-Temaimi, Tuan Zea Tan, Makia J. Marafie, Jean Paul Thiery, Philip Quirke and Fahd Al-Mulla Figure S1. Cont. Int. J. Mol. Sci. 2016, 17, 598; doi:10.3390/ijms17040598 S2 of S16 Figure S1. Mean expression levels of fourteen genes of significant association with CRC DFS and OS that are differentially expressed in normal colon compared to CRC tissues. Each dot represents a sample. Table S1. Copy number aberrations associated with poor disease-free survival and metastasis in early stage II CRC as predicted by STAC and SPPS combined methodologies with resident gene symbols. CN stands for copy number, whereas CNV is copy number variation. Region Cytoband % of CNV Count of Region Event Gene Symbols Length Location Overlap Genes chr1:113,025,076–113,199,133 174,057 p13.2 CN Loss 0.0 2 AKR7A2P1, SLC16A1 chr1:141,465,960–141,822,265 356,305 q12–q21.1 CN Gain 95.9 1 SRGAP2B MIR5087, LOC10013000 0, FLJ39739, LOC10028679 3, PPIAL4G, PPIAL4A, NBPF14, chr1:144,911,564–146,242,907 1,331,343 q21.1 CN Gain 99.6 16 NBPF15, NBPF16, PPIAL4E, NBPF16, PPIAL4D, PPIAL4F, LOC645166, LOC388692, FCGR1C chr1:177,209,428–177,226,812 17,384 q25.3 CN Gain 0.0 0 chr1:197,652,888–197,676,831 23,943 q32.1 CN Gain 0.0 1 KIF21B chr1:201,015,278–201,033,308 18,030 q32.1 CN Gain 0.0 1 PLEKHA6 chr1:201,289,154–201,298,247 9093 q32.1 CN Gain 0.0 0 chr1:216,820,186–217,043,421 223,235 q41 CN -

Termination of RNA Polymerase II Transcription by the 5’-3’ Exonuclease Xrn2
TERMINATION OF RNA POLYMERASE II TRANSCRIPTION BY THE 5’-3’ EXONUCLEASE XRN2 by MICHAEL ANDRES CORTAZAR OSORIO B.S., Universidad del Valle – Colombia, 2011 A thesis submitted to the Faculty of the Graduate School of the University of Colorado in partial fulfillment of the requirements for the degree of Doctor of Philosophy Molecular Biology Program 2018 This thesis for the Doctor of Philosophy degree by Michael Andrés Cortázar Osorio has been approved for the Molecular Biology Program by Mair Churchill, Chair Richard Davis Jay Hesselberth Thomas Blumenthal James Goodrich David Bentley, Advisor Date: Aug 17, 2018 ii Cortázar Osorio, Michael Andrés (Ph.D., Molecular Biology) Termination of RNA polymerase II transcription by the 5’-3’ exonuclease Xrn2 Thesis directed by Professor David L. Bentley ABSTRACT Termination of transcription occurs when RNA polymerase (pol) II dissociates from the DNA template and releases a newly-made mRNA molecule. Interestingly, an active debate fueled by conflicting reports over the last three decades is still open on which of the two main models of termination of RNA polymerase II transcription does in fact operate at 3’ ends of genes. The torpedo model indicates that the 5’-3’ exonuclease Xrn2 targets the nascent transcript for degradation after cleavage at the polyA site and chases pol II for termination. In contrast, the allosteric model asserts that transcription through the polyA signal induces a conformational change of the elongation complex and converts it into a termination-competent complex. In this thesis, I propose a unified allosteric-torpedo mechanism. Consistent with a polyA site-dependent conformational change of the elongation complex, I found that pol II transitions at the polyA site into a mode of slow transcription elongation that is accompanied by loss of Spt5 phosphorylation in the elongation complex. -

Fibrosis Accumulation in Idiopathic Pulmonary Apoptosis
Increased Cell Surface Fas Expression Is Necessary and Sufficient To Sensitize Lung Fibroblasts to Fas Ligation-Induced Apoptosis: Implications for Fibroblast This information is current as Accumulation in Idiopathic Pulmonary of October 1, 2021. Fibrosis Murry W. Wynes, Benjamin L. Edelman, Amanda G. Kostyk, Michael G. Edwards, Christopher Coldren, Steve D. Groshong, Gregory P. Cosgrove, Elizabeth F. Redente, Alison Bamberg, Kevin K. Brown, Nichole Reisdorph, Downloaded from Rebecca C. Keith, Stephen K. Frankel and David W. H. Riches J Immunol 2011; 187:527-537; Prepublished online 1 June 2011; doi: 10.4049/jimmunol.1100447 http://www.jimmunol.org/ http://www.jimmunol.org/content/187/1/527 Supplementary http://www.jimmunol.org/content/suppl/2011/06/01/jimmunol.110044 Material 7.DC1 References This article cites 62 articles, 18 of which you can access for free at: by guest on October 1, 2021 http://www.jimmunol.org/content/187/1/527.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2011 by The American Association of Immunologists, Inc. -

Expansions of Adaptive-Like NK Cells with a Tissue-Resident Phenotype in Human Lung and Blood
Expansions of adaptive-like NK cells with a tissue-resident phenotype in human lung and blood Demi Brownliea,1, Marlena Scharenberga,1, Jeff E. Moldb, Joanna Hårdb, Eliisa Kekäläinenc,d,e, Marcus Buggerta, Son Nguyenf,g, Jennifer N. Wilsona, Mamdoh Al-Amerih, Hans-Gustaf Ljunggrena, Nicole Marquardta,2,3, and Jakob Michaëlssona,2 aCenter for Infectious Medicine, Department of Medicine Huddinge, Karolinska Institutet, 14152 Stockholm, Sweden; bDepartment of Cell and Molecular Biology, Karolinska Institutet, 171 77 Stockholm, Sweden; cTranslational Immunology Research Program, University of Helsinki, 00014 Helsinki, Finland; dDepartment of Bacteriology and Immunology, University of Helsinki, 00014 Helsinki, Finland; eHelsinki University Central Hospital Laboratory, Division of Clinical Microbiology, Helsinki University Hospital, 00290 Helsinki, Finland; fDepartment of Microbiology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104; gInstitute for Immunology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104; and hThoracic Surgery, Department of Molecular Medicine and Surgery, Karolinska University Hospital, Karolinska Institutet, 171 76 Stockholm, Sweden Edited by Marco Colonna, Washington University in St. Louis School of Medicine, St. Louis, MO, and approved January 27, 2021 (received for review August 18, 2020) Human adaptive-like “memory” CD56dimCD16+ natural killer (NK) We and others recently identified a subset of tissue-resident − cells in peripheral blood from cytomegalovirus-seropositive indi- CD49a+CD56brightCD16 NK cells in the human lung (14, 15). viduals have been extensively investigated in recent years and are The human lung is a frequent site of infection with viruses such currently explored as a treatment strategy for hematological can- as influenza virus and HCMV, as well as a reservoir for latent cers. -

Exploring the Regulatory Mechanism of the Notch Ligand Receptor Jagged1 Via the Aryl Hydrocarbon Receptor in Breast Cancer Sean Alan Piwarski
Marshall University Marshall Digital Scholar Theses, Dissertations and Capstones 2018 Exploring the Regulatory Mechanism of the Notch Ligand Receptor Jagged1 Via the Aryl Hydrocarbon Receptor in Breast Cancer Sean Alan Piwarski Follow this and additional works at: https://mds.marshall.edu/etd Part of the Biological Phenomena, Cell Phenomena, and Immunity Commons, Medical Molecular Biology Commons, Medical Toxicology Commons, and the Oncology Commons EXPLORING THE REGULATORY MECHANISM OF THE NOTCH LIGAND RECEPTOR JAGGED1 VIA THE ARYL HYDROCARBON RECEPTOR IN BREAST CANCER A dissertation submitted to the Graduate College of Marshall University In partial fulfillment of the requirements for the degree of Doctor of Philosophy In Biomedical Sciences by Sean Alan Piwarski Approved by Dr. Travis Salisbury, Committee Chairperson Dr. Gary Rankin Dr. Monica Valentovic Dr. Richard Egleton Dr. Todd Green Marshall University July 2018 APPROVAL OF DISSERTATION We, the faculty supervising the work of Sean Alan Piwarski, affirm that the dissertation, Exploring the Regulatory Mechanism of the Notch Ligand Receptor JAGGED1 via the Aryl Hydrocarbon Receptor in Breast Cancer, meets the high academic standards for original scholarship and creative work established by the Biomedical Sciences program and the Graduate College of Marshall University. This work also conforms to the editorial standards of our discipline and the Graduate College of Marshall University. With our signatures, we approve the manuscript for publication. ii © 2018 SEAN ALAN PIWARSKI ALL RIGHTS RESERVED iii DEDICATION To my mom and dad, This dissertation is a product of your hard work and sacrifice as amazing parents. I could never have accomplished something of this magnitude without your love, support, and constant encouragement. -

PDF Datasheet
Product Datasheet C15orf39 Antibody NBP1-90552 Unit Size: 0.1 ml Store at 4C short term. Aliquot and store at -20C long term. Avoid freeze-thaw cycles. Protocols, Publications, Related Products, Reviews, Research Tools and Images at: www.novusbio.com/NBP1-90552 Updated 9/16/2020 v.20.1 Earn rewards for product reviews and publications. Submit a publication at www.novusbio.com/publications Submit a review at www.novusbio.com/reviews/destination/NBP1-90552 Page 1 of 3 v.20.1 Updated 9/16/2020 NBP1-90552 C15orf39 Antibody Product Information Unit Size 0.1 ml Concentration Concentrations vary lot to lot. See vial label for concentration. If unlisted please contact technical services. Storage Store at 4C short term. Aliquot and store at -20C long term. Avoid freeze-thaw cycles. Clonality Polyclonal Preservative 0.02% Sodium Azide Isotype IgG Purity Immunogen affinity purified Buffer PBS (pH 7.2) and 40% Glycerol Product Description Host Rabbit Gene ID 56905 Gene Symbol C15ORF39 Species Human Reactivity Notes Immunogen displays the following percentage of sequence identity for non- tested species: Rat (89%) Specificity/Sensitivity Specificity of human C15orf39 antibody verified on a Protein Array containing target protein plus 383 other non-specific proteins. Immunogen This antibody was developed against Recombinant Protein corresponding to amino acids: LCDAISGSVAHSPPEKLREWLETAGPWGQAAWQDCQGVQGLLAKLLSQLQRF DRTHRCPFPHVVRAGAIFVPIHLVKERLFPRLPPASVDHVL Product Application Details Applications Immunocytochemistry/Immunofluorescence, Immunohistochemistry, Immunohistochemistry-Paraffin Recommended Dilutions Immunohistochemistry 1:200 - 1:500, Immunocytochemistry/Immunofluorescence 0.25-2 ug/ml, Immunohistochemistry-Paraffin 1:200 - 1:500 Application Notes For IHC-Paraffin, HIER pH 6 retrieval is recommended. Immunocytochemistry/Immunofluorescence Fixation Permeabilization: Use PFA/Triton X-100. -

Molecular Regulation of Gliogenesis in Xenopus Laevis During Primary
Molecular Regulation of Gliogenesis in Xenopus laevis during Primary Neurogenesis ------------------------------------------------------ A Dissertation Presented to the Faculty of the Department of Biology and Biochemistry University of Houston ------------------------------------------------------ In Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy ------------------------------------------------------ By Christina Helen Ulrich August 2019 Molecular Regulation of Gliogenesis in Xenopus laevis during Primary Neurogenesis ______________________________ Christina H. Ulrich APPROVED: ______________________________ Dr. Amy K. Sater, Chair ______________________________ Dr. Brigitte Dauwalder ______________________________ Dr. Arne Lekven ______________________________ Dr. Rachel K. Miller ______________________________ Dean, College of Natural Sciences and Mathematics ii In memory of my mom, and for my dad. iii Acknowledgements First, I would like to thank my mentor and friend, Dr. Amy K. Sater. She has provided countless hours of teaching and feedback and without her continual support and encouragement I could not have completed my studies. I would also like to thank my committee members, Dr. Brigitte Dauwalder, Dr. Rachel K. Miller, and Dr. Arne Lekven. They have provided valuable insight and asked questions that opened new avenues of thinking in my project. I would also like to recognize Dr. Robert Schwartz, he provided support and encouragement during difficult periods of my graduate studies. I am grateful to my family; my parents, Nancy and Sam Ulrich, and my siblings, Samantha, Marta, and Ben, who patiently listened to me practice my science communication on them. Without their love and support, I would never have started or finished graduate school. I want to thank my incredibly supportive partner Jon, his love of science and unflagging enthusiasm offsets my more pessimistic view of the world. -

Mechanism of Ant1-Induced Human Diseases and Stress Signaling
MECHANISM OF ANT1-INDUCED HUMAN DISEASES AND STRESS SIGNALING Item Type Dissertation Authors Liu, Yaxin Rights Attribution-NonCommercial-NoDerivatives 4.0 International Download date 09/10/2021 19:50:21 Item License http://creativecommons.org/licenses/by-nc-nd/4.0/ Link to Item http://hdl.handle.net/20.500.12648/1772 MECHANISM OF ANT1-INDUCED HUMAN DISEASES AND STRESS SIGNALING Yaxin Liu A Dissertation in Department of Biochemistry and Molecular Biology Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the College of Graduate Studies of State University of New York, Upstate Medical University. Approved Date Abstract Mechanism of Ant1-induced Human Diseases and Stress Signaling Yaxin Liu Xin Jie Chen Adenine nucleotide translocase (Ant) is a mitochondrial inner membrane protein, the primary function of which is to mediate the ADP/ATP exchange across the inner membrane. Missense mutations in Ant1, the skeletal muscle- and heart-specific isoform, induce human disorders including autosomal dominant Progressive External Opthalmoplegia (adPEO), cardiomyopathy and myopathy. Several models were proposed to interpret the pathogenesis of mutant Ant1-induced diseases, but no consensus has been reached. Our lab has previously found that mutant Aac2, the homolog of Ant1 in yeast, causes cell death due to the mitochondrial biogenesis defect. In the present study, we provided biochemical evidence supporting the idea that the mutant Aac2 proteins are misfolded, which derails the proteostasis on the inner membrane. We found that the assembly and stability of multiple protein complexes on the inner membrane are affected, including those involved in mitochondrial respiration and protein transport. -

Autocrine IFN Signaling Inducing Profibrotic Fibroblast Responses By
Downloaded from http://www.jimmunol.org/ by guest on September 23, 2021 Inducing is online at: average * The Journal of Immunology , 11 of which you can access for free at: 2013; 191:2956-2966; Prepublished online 16 from submission to initial decision 4 weeks from acceptance to publication August 2013; doi: 10.4049/jimmunol.1300376 http://www.jimmunol.org/content/191/6/2956 A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Autocrine IFN Signaling Feng Fang, Kohtaro Ooka, Xiaoyong Sun, Ruchi Shah, Swati Bhattacharyya, Jun Wei and John Varga J Immunol cites 49 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130037 6.DC1 This article http://www.jimmunol.org/content/191/6/2956.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 23, 2021. The Journal of Immunology A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Inducing Autocrine IFN Signaling Feng Fang,* Kohtaro Ooka,* Xiaoyong Sun,† Ruchi Shah,* Swati Bhattacharyya,* Jun Wei,* and John Varga* Activation of TLR3 by exogenous microbial ligands or endogenous injury-associated ligands leads to production of type I IFN. -

GENE LIST ANTI-CORRELATED Systematic Common Description
GENE LIST ANTI-CORRELATED Systematic Common Description 210348_at 4-Sep Septin 4 206155_at ABCC2 ATP-binding cassette, sub-family C (CFTR/MRP), member 2 221226_s_at ACCN4 Amiloride-sensitive cation channel 4, pituitary 207427_at ACR Acrosin 214957_at ACTL8 Actin-like 8 207422_at ADAM20 A disintegrin and metalloproteinase domain 20 216998_s_at ADAM5 synonym: tMDCII; Homo sapiens a disintegrin and metalloproteinase domain 5 (ADAM5) on chromosome 8. 216743_at ADCY6 Adenylate cyclase 6 206807_s_at ADD2 Adducin 2 (beta) 208544_at ADRA2B Adrenergic, alpha-2B-, receptor 38447_at ADRBK1 Adrenergic, beta, receptor kinase 1 219977_at AIPL1 211560_s_at ALAS2 Aminolevulinate, delta-, synthase 2 (sideroblastic/hypochromic anemia) 211004_s_at ALDH3B1 Aldehyde dehydrogenase 3 family, member B1 204705_x_at ALDOB Aldolase B, fructose-bisphosphate 220365_at ALLC Allantoicase 204664_at ALPP Alkaline phosphatase, placental (Regan isozyme) 216377_x_at ALPPL2 Alkaline phosphatase, placental-like 2 221114_at AMBN Ameloblastin, enamel matrix protein 206892_at AMHR2 Anti-Mullerian hormone receptor, type II 217293_at ANGPT1 Angiopoietin 1 210952_at AP4S1 Adaptor-related protein complex 4, sigma 1 subunit 207158_at APOBEC1 Apolipoprotein B mRNA editing enzyme, catalytic polypeptide 1 213611_at AQP5 Aquaporin 5 216219_at AQP6 Aquaporin 6, kidney specific 206784_at AQP8 Aquaporin 8 214490_at ARSF Arylsulfatase F 216204_at ARVCF Armadillo repeat gene deletes in velocardiofacial syndrome 214070_s_at ATP10B ATPase, Class V, type 10B 221240_s_at B3GNT4 UDP-GlcNAc:betaGal -

Exploring the Genetic Associations Between the Use of Statins and Alzheimer's Disease
J Lipid Atheroscler. 2021 Sep;10(3):e27 Journal of https://doi.org/10.12997/jla.2021.10.e27 Lipid and pISSN 2287-2892·eISSN 2288-2561 Atherosclerosis Original Article Exploring the Genetic Associations between the Use of Statins and Alzheimer's Disease Jibeom Lee ,1 Suhyeon Park ,1 Yumin Kim ,1 Hyun Min Kim ,2 Chang-Myung Oh 1 1Department of Biomedical Science and Engineering, Gwangju Institute of Science and Technology, Gwangju, Korea 2Division of Endocrinology and Metabolism, Department of Internal Medicine, Chung-Ang University College of Medicine, Seoul, Korea Received: Mar 13, 2021 Revised: May 10, 2021 ABSTRACT Accepted: May 18, 2021 Objective: Alzheimer's disease (AD) is the most common cause of dementia. The statins Correspondence to have shown beneficial effects on cognitive functions and reduced the risk of dementia Hyun Min Kim development. However, the exact mechanisms of statin effects in AD are not yet fully Division of Endocrinology and Metabolism, Department of Internal Medicine, Chung-Ang understood. In this study, we aimed to explore the underlying mechanisms of statin on AD. University College of Medicine, 84 Heukseok- Methods: We downloaded AD blood dataset (GSE63060) and statin-related blood gene ro, Dongjak-gu, Seoul 06974, Korea. expression dataset (GSE86216). Then we performed gene expression analysis of each dataset E-mail: [email protected] and compared blood gene expressions between AD patients and statin-treated patients. Chang-Myung Oh Then, we downloaded mouse embryonic neural stem cell dataset (GSE111945) and performed Department of Biomedical Science and gene expression analysis. Engineering, Gwangju Institute of Science and Results: From the human blood dataset, we identified upregulated/downregulated genes Technology, 123 Cheomdangwagi-ro, Buk-gu, in AD patients and statin-treated patients.