Crystal Structure of a Hidden Protein, Ycac, a Putative Cysteine Hydrolase from Pseudomonas Aeruginosa, with and Without an Acrylamide Adduct
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Downloaded 10/5/2021 9:43:05 AM
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue Aromatic side-chain flips orchestrate the conformational sampling of functional loops in Cite this: Chem. Sci.,2021,12,9318 † All publication charges for this article human histone deacetylase 8 have been paid for by the Royal Society a a bcd of Chemistry Vaibhav Kumar Shukla, ‡ Lucas Siemons, ‡ Francesco L. Gervasio and D. Flemming Hansen *a Human histone deacetylase 8 (HDAC8) is a key hydrolase in gene regulation and an important drug-target. High-resolution structures of HDAC8 in complex with substrates or inhibitors are available, which have provided insights into the bound state of HDAC8 and its function. Here, using long all-atom unbiased molecular dynamics simulations and Markov state modelling, we show a strong correlation between the conformation of aromatic side chains near the active site and opening and closing of the surrounding functional loops of HDAC8. We also investigated two mutants known to allosterically downregulate the enzymatic activity of HDAC8. Based on experimental data, we hypothesise that I19S-HDAC8 is unable to Received 6th April 2021 Creative Commons Attribution 3.0 Unported Licence. release the product, whereas both product release and substrate binding are impaired in the S39E- Accepted 27th May 2021 HDAC8 mutant. The presented results deliver detailed insights into the functional dynamics of HDAC8 DOI: 10.1039/d1sc01929e and provide a mechanism for the substantial downregulation caused by allosteric mutations, including rsc.li/chemical-science a disease causing one. Introduction II (HDAC-4, -5, -6, -7, -9, and HDAC10), and class III (SIRT1-7) have sequence similarity to yeast Rpd3, Hda1, and Sir2, Acetylation of lysine side chains occurs as a co-translation or respectively, whereas class IV (HDAC11) shares sequence simi- 2 This article is licensed under a post-translational modication of proteins and was rst iden- larity with both class I and II proteins. -

1 Metabolic Dysfunction Is Restricted to the Sciatic Nerve in Experimental
Page 1 of 255 Diabetes Metabolic dysfunction is restricted to the sciatic nerve in experimental diabetic neuropathy Oliver J. Freeman1,2, Richard D. Unwin2,3, Andrew W. Dowsey2,3, Paul Begley2,3, Sumia Ali1, Katherine A. Hollywood2,3, Nitin Rustogi2,3, Rasmus S. Petersen1, Warwick B. Dunn2,3†, Garth J.S. Cooper2,3,4,5* & Natalie J. Gardiner1* 1 Faculty of Life Sciences, University of Manchester, UK 2 Centre for Advanced Discovery and Experimental Therapeutics (CADET), Central Manchester University Hospitals NHS Foundation Trust, Manchester Academic Health Sciences Centre, Manchester, UK 3 Centre for Endocrinology and Diabetes, Institute of Human Development, Faculty of Medical and Human Sciences, University of Manchester, UK 4 School of Biological Sciences, University of Auckland, New Zealand 5 Department of Pharmacology, Medical Sciences Division, University of Oxford, UK † Present address: School of Biosciences, University of Birmingham, UK *Joint corresponding authors: Natalie J. Gardiner and Garth J.S. Cooper Email: [email protected]; [email protected] Address: University of Manchester, AV Hill Building, Oxford Road, Manchester, M13 9PT, United Kingdom Telephone: +44 161 275 5768; +44 161 701 0240 Word count: 4,490 Number of tables: 1, Number of figures: 6 Running title: Metabolic dysfunction in diabetic neuropathy 1 Diabetes Publish Ahead of Print, published online October 15, 2015 Diabetes Page 2 of 255 Abstract High glucose levels in the peripheral nervous system (PNS) have been implicated in the pathogenesis of diabetic neuropathy (DN). However our understanding of the molecular mechanisms which cause the marked distal pathology is incomplete. Here we performed a comprehensive, system-wide analysis of the PNS of a rodent model of DN. -

Mechanistic Implications for the Chorismatase Fkbo Based on the Crystal Structure
Mechanistic Implications for the Chorismatase FkbO Based on the Crystal Structure Puneet Juneja 1,†, Florian Hubrich 2,†, Kay Diederichs 1, Wolfram Welte 1 and Jennifer N. Andexer 2 1 - Department of Biology, University of Konstanz, D-78457 Konstanz, Germany 2 - Institute of Pharmaceutical Sciences, Albert-Ludwigs-University Freiburg, Albertstr 25, D-79104 Freiburg, Germany Correspondence to Jennifer N. Andexer: [email protected] http://dx.doi.org/10.1016/j.jmb.2013.09.006 Edited by M. Guss Abstract Chorismate-converting enzymes are involved in many biosynthetic pathways leading to natural products and can often be used as tools for the synthesis of chemical building blocks. Chorismatases such as FkbO from Streptomyces species catalyse the hydrolysis of chorismate yielding (dihydro)benzoic acid derivatives. In contrast to many other chorismate-converting enzymes, the structure and catalytic mechanism of a chorismatase had not been previously elucidated. Here we present the crystal structure of the chorismatase FkbO in complex with a competitive inhibitor at 1.08 Å resolution. FkbO is a monomer in solution and exhibits pseudo-3-fold symmetry; the structure of the individual domains indicates a possible connection to the trimeric RidA/YjgF family and related enzymes. The co-crystallised inhibitor led to the identification of FkbO's active site in the cleft between the central and the C-terminal domains. A mechanism for FkbO is proposed based on both interactions between the inhibitor and the surrounding amino acids and an FkbO structure with chorismate modelled in the active site. We suggest that the methylene group of the chorismate enol ether takes up a proton from an active-site glutamic acid residue, thereby initiating chorismate hydrolysis. -
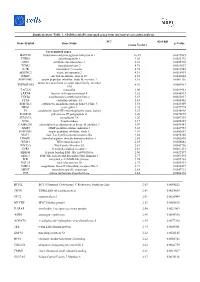
Supplementary Table 1. All Differentially Expressed Genes from Microarray Screening Analysis
Supplementary Table 1. All differentially expressed genes from microarray screening analysis. FCa (Gal-KD Gene Symbol Gene Name p-Value versus Vector) Up-regulated genes HAPLN1 hyaluronan and proteoglycan link protein 1 10.49 0.0027085 THBS1 thrombospondin 1 9.20 0.0022192 ODC1 ornithine decarboxylase 1 6.38 0.0055776 TGM2 transglutaminase 2 4.76 0.0015627 IL7R interleukin 7 receptor 4.75 0.0017245 SERINC2 serine incorporator 2 4.51 0.0014919 ITM2C integral membrane protein 2C 4.32 0.0044644 SERPINB7 serpin peptidase inhibitor, clade B, member 7 4.18 0.0081136 tumor necrosis factor receptor superfamily, member TNFRSF10D 4.01 0.0085561 10d TAGLN transgelin 3.90 0.0099963 LRRN4 leucine rich repeat neuronal 4 3.82 0.0046513 TGFB2 transforming growth factor beta 2 3.51 0.0035017 CPA4 carboxypeptidase A4 3.43 0.0008452 EPB41L3 erythrocyte membrane protein band 4.1-like 3 3.34 0.0025309 NRG1 neuregulin 1 3.28 0.0079724 F3 coagulation factor III (thromboplastin, tissue factor) 3.27 0.0038968 POLR3G polymerase III polypeptide G 3.26 0.0070675 SEMA7A semaphorin 7A 3.20 0.0087335 NT5E 5-nucleotidase 3.17 0.0036353 CAMK2N1 calmodulin-dependent protein kinase II inhibitor 1 3.07 0.0090141 TIMP3 TIMP metallopeptidase inhibitor 3 3.03 0.0047953 SERPINE1 serpin peptidase inhibitor, clade E 2.97 0.0053652 MALL mal, T-cell differentiation protein-like 2.88 0.0078205 DDAH1 dimethylarginine dimethylaminohydrolase 1 2.86 0.0002895 WDR3 WD repeat domain 3 2.85 0.0058842 WNT5A Wnt Family Member 5A 2.81 0.0043796 GPR1 G protein-coupled receptor 1 2.81 0.0021313 -

Phza/B Catalyzes the Formation of the Tricycle in Phenazine Biosynthesis Ekta G
Subscriber access provided by DigiTop | USDA's Digital Desktop Library Article PhzA/B Catalyzes the Formation of the Tricycle in Phenazine Biosynthesis Ekta G. Ahuja, Petra Janning, Matthias Mentel, Almut Graebsch, Rolf Breinbauer, Wolf Hiller, Burkhard Costisella, Linda S. Thomashow, Dmitri V. Mavrodi, and Wulf Blankenfeldt J. Am. Chem. Soc., 2008, 130 (50), 17053-17061 • DOI: 10.1021/ja806325k • Publication Date (Web): 17 November 2008 Downloaded from http://pubs.acs.org on January 15, 2009 More About This Article Additional resources and features associated with this article are available within the HTML version: • Supporting Information • Access to high resolution figures • Links to articles and content related to this article • Copyright permission to reproduce figures and/or text from this article Journal of the American Chemical Society is published by the American Chemical Society. 1155 Sixteenth Street N.W., Washington, DC 20036 Published on Web 11/17/2008 PhzA/B Catalyzes the Formation of the Tricycle in Phenazine Biosynthesis Ekta G. Ahuja,† Petra Janning,† Matthias Mentel,‡,§ Almut Graebsch,‡ Rolf Breinbauer,†,‡,§,| Wolf Hiller,‡ Burkhard Costisella,‡ Linda S. Thomashow,⊥,# Dmitri V. Mavrodi,⊥ and Wulf Blankenfeldt*,† Max-Planck-Institute of Molecular Physiology, Otto-Hahn-Strasse 11, 44227 Dortmund, Germany, Technical UniVersity of Dortmund, Faculty of Chemistry, Otto-Hahn-Strasse 6, 44221 Dortmund, Germany, UniVersity of Leipzig, Institute of Organic Chemistry, Johannisallee 29, 04103 Leipzig, Germany, Graz UniVersity of -

Biochemical Characterization and Comparison of Aspartylglucosaminidases Secreted in Venom of the Parasitoid Wasps Asobara Tabida and Leptopilina Heterotoma
RESEARCH ARTICLE Biochemical characterization and comparison of aspartylglucosaminidases secreted in venom of the parasitoid wasps Asobara tabida and Leptopilina heterotoma Quentin Coulette1, SeÂverine Lemauf2, Dominique Colinet2, Geneviève PreÂvost1, Caroline Anselme1, Marylène Poirie 2, Jean-Luc Gatti2* a1111111111 1 Unite ªEcologie et Dynamique des Systèmes AnthropiseÂsº (EDYSAN, FRE 3498 CNRS-UPJV), Universite de Picardie Jules Verne, Amiens, France, 2 Universite CoÃte d'Azur, INRA, CNRS, ISA, Sophia Antipolis, a1111111111 France a1111111111 a1111111111 * [email protected] a1111111111 Abstract Aspartylglucosaminidase (AGA) is a low-abundance intracellular enzyme that plays a key OPEN ACCESS role in the last stage of glycoproteins degradation, and whose deficiency leads to human Citation: Coulette Q, Lemauf S, Colinet D, PreÂvost aspartylglucosaminuria, a lysosomal storage disease. Surprisingly, high amounts of AGA- G, Anselme C, Poirie M, et al. (2017) Biochemical characterization and comparison of like proteins are secreted in the venom of two phylogenetically distant hymenopteran para- aspartylglucosaminidases secreted in venom of the sitoid wasp species, Asobara tabida (Braconidae) and Leptopilina heterotoma (Cynipidae). parasitoid wasps Asobara tabida and Leptopilina These venom AGAs have a similar domain organization as mammalian AGAs. They share heterotoma. PLoS ONE 12(7): e0181940. https:// with them key residues for autocatalysis and activity, and the mature - and -subunits also doi.org/10.1371/journal.pone.0181940 α β form an (αβ)2 structure in solution. Interestingly, only one of these AGAs subunits (α for Editor: Erjun Ling, Institute of Plant Physiology and AtAGA and for LhAGA) is glycosylated instead of the two subunits for lysosomal human Ecology Shanghai Institutes for Biological β Sciences, CHINA AGA (hAGA), and these glycosylations are partially resistant to PGNase F treatment. -

Chorismate Mutase and Isochorismatase, Two Potential
Received: 28 June 2020 | Revised: 7 September 2020 | Accepted: 7 September 2020 DOI: 10.1111/mpp.13003 ORIGINAL ARTICLE Chorismate mutase and isochorismatase, two potential effectors of the migratory nematode Hirschmanniella oryzae, increase host susceptibility by manipulating secondary metabolite content of rice Lander Bauters 1 | Tina Kyndt 1 | Tim De Meyer2 | Kris Morreel3,4 | Wout Boerjan3,4 | Hannes Lefevere1 | Godelieve Gheysen 1 1Department of Biotechnology, Faculty of Bioscience Engineering, Ghent University, Abstract Ghent, Belgium Hirschmanniella oryzae is one of the most devastating nematodes on rice, leading to 2 Department of Data Analysis and substantial yield losses. Effector proteins aid the nematode during the infection pro- Mathematical Modelling, Faculty of Bioscience Engineering, Ghent University, cess by subduing plant defence responses. In this research we characterized two po- Ghent, Belgium tential H. oryzae effector proteins, chorismate mutase (HoCM) and isochorismatase 3VIB-UGent Center for Plant Systems Biology, Ghent, Belgium (HoICM), and investigated their enzymatic activity and their role in plant immunity. 4Department of Plant Biotechnology and Both HoCM and HoICM proved to be enzymatically active in complementation tests Bioinformatics, Faculty of Sciences, Ghent in mutant Escherichia coli strains. Infection success by the migratory nematode H. ory- University, Ghent, Belgium zae was significantly higher in transgenic rice lines constitutively expressing HoCM Correspondence or HoICM. Expression of HoCM, but not HoICM, increased rice susceptibility against Godelieve Gheysen, Department of Biotechnology, Faculty of Bioscience the sedentary nematode Meloidogyne graminicola also. Transcriptome and metabo- Engineering, Ghent University, Ghent, lome analyses indicated reductions in secondary metabolites in the transgenic rice Belgium. Email: [email protected] plants expressing the potential nematode effectors. -

Biosynthesis of Natural Products Containing Β-Amino Acids
Natural Product Reports Biosynthesis of natural products containing β -amino acids Journal: Natural Product Reports Manuscript ID: NP-REV-01-2014-000007.R1 Article Type: Review Article Date Submitted by the Author: 21-Apr-2014 Complete List of Authors: Kudo, Fumitaka; Tokyo Institute Of Technology, Department of Chemistry Miyanaga, Akimasa; Tokyo Institute Of Technology, Department of Chemistry Eguchi, T; Tokyo Institute Of Technology, Department of Chemistry and Materials Science Page 1 of 20 Natural Product Reports NPR RSC Publishing REVIEW Biosynthesis of natural products containing βββ- amino acids Cite this: DOI: 10.1039/x0xx00000x Fumitaka Kudo, a Akimasa Miyanaga, a and Tadashi Eguchi *b Received 00th January 2014, We focus here on β-amino acids as components of complex natural products because the presence of β-amino acids Accepted 00th January 2014 produces structural diversity in natural products and provides characteristic architectures beyond that of ordinary DOI: 10.1039/x0xx00000x α-L-amino acids, thus generating significant and unique biological functions in nature. In this review, we first survey the known bioactive β-amino acid-containing natural products including nonribosomal peptides, www.rsc.org/ macrolactam polyketides, and nucleoside-β-amino acid hybrids. Next, the biosynthetic enzymes that form β-amino acids from α-amino acids and de novo synthesis of β-amino acids are summarized. Then, the mechanisms of β- amino acid incorporation into natural products are reviewed. Because it is anticipated that the rational swapping of the β-amino acid moieties with various side chains and stereochemistries by biosynthetic engineering should lead to the creation of novel architectures and bioactive compounds, the accumulation of knowledge regarding β- amino acid-containing natural product biosynthetic machinery could have a significant impact in this field. -

Active Site Tyrosine Is Essential for Amidohydrolase but Not for Esterase
Active site tyrosine is essential for amidohydrolase but not for esterase activity of a class 2 histone deacetylase-like bacterial enzyme Kristin Moreth, Daniel Riester, Christian Hildmann, René Hempel, Dennis Wegener, Andreas Schober, Andreas Schwienhorst To cite this version: Kristin Moreth, Daniel Riester, Christian Hildmann, René Hempel, Dennis Wegener, et al.. Ac- tive site tyrosine is essential for amidohydrolase but not for esterase activity of a class 2 histone deacetylase-like bacterial enzyme. Biochemical Journal, Portland Press, 2006, 401 (3), pp.659-665. 10.1042/BJ20061239. hal-00478649 HAL Id: hal-00478649 https://hal.archives-ouvertes.fr/hal-00478649 Submitted on 30 Apr 2010 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Biochemical Journal Immediate Publication. Published on 12 Oct 2006 as manuscript BJ20061239 Active site tyrosine is essential for amidohydrolase but not for esterase activity of a class 2 histone deacetylase-like bacterial enzyme Kristin Moreth¶, Daniel Riester¶, Christian Hildmann¶, René Hempel¶, Dennis Wegener¶,§,‡, -

NIH Public Access Author Manuscript Arch Biochem Biophys
NIH Public Access Author Manuscript Arch Biochem Biophys. Author manuscript; available in PMC 2014 November 01. NIH-PA Author ManuscriptPublished NIH-PA Author Manuscript in final edited NIH-PA Author Manuscript form as: Arch Biochem Biophys. 2013 November 1; 539(1): . doi:10.1016/j.abb.2013.09.007. Redesign of MST enzymes to target lyase activity instead promotes mutase and dehydratase activities Kathleen M. Meneely, Qianyi Luo, and Audrey L. Lamb* Department of Molecular Biosciences, University of Kansas, Lawrence, Kansas 66045 Abstract The isochorismate and salicylate synthases are members of the MST family of enzymes. The isochorismate synthases establish an equilibrium for the conversion chorismate to isochorismate and the reverse reaction. The salicylate synthases convert chorismate to salicylate with an isochorismate intermediate; therefore, the salicylate synthases perform isochorismate synthase and isochorismate-pyruvate lyase activities sequentially. While the active site residues are highly conserved, there are two sites that show trends for lyase-activity and lyase-deficiency. Using steady state kinetics and HPLC progress curves, we tested the “interchange” hypothesis that interconversion of the amino acids at these sites would promote lyase activity in the isochorismate synthases and remove lyase activity from the salicylate synthases. An alternative, “permute” hypothesis, that chorismate-utilizing enzymes are designed to permute the substrate into a variety of products and tampering with the active site may lead to identification of adventitious activities, is tested by more sensitive NMR time course experiments. The latter hypothesis held true. The variant enzymes predominantly catalyzed chorismate mutase-prephenate dehydratase activities, sequentially generating prephenate and phenylpyruvate, augmenting previously debated (mutase) or undocumented (dehydratase) adventitious activities. -

Supplementary Informations SI2. Supplementary Table 1
Supplementary Informations SI2. Supplementary Table 1. M9, soil, and rhizosphere media composition. LB in Compound Name Exchange Reaction LB in soil LBin M9 rhizosphere H2O EX_cpd00001_e0 -15 -15 -10 O2 EX_cpd00007_e0 -15 -15 -10 Phosphate EX_cpd00009_e0 -15 -15 -10 CO2 EX_cpd00011_e0 -15 -15 0 Ammonia EX_cpd00013_e0 -7.5 -7.5 -10 L-glutamate EX_cpd00023_e0 0 -0.0283302 0 D-glucose EX_cpd00027_e0 -0.61972444 -0.04098397 0 Mn2 EX_cpd00030_e0 -15 -15 -10 Glycine EX_cpd00033_e0 -0.0068175 -0.00693094 0 Zn2 EX_cpd00034_e0 -15 -15 -10 L-alanine EX_cpd00035_e0 -0.02780553 -0.00823049 0 Succinate EX_cpd00036_e0 -0.0056245 -0.12240603 0 L-lysine EX_cpd00039_e0 0 -10 0 L-aspartate EX_cpd00041_e0 0 -0.03205557 0 Sulfate EX_cpd00048_e0 -15 -15 -10 L-arginine EX_cpd00051_e0 -0.0068175 -0.00948672 0 L-serine EX_cpd00054_e0 0 -0.01004986 0 Cu2+ EX_cpd00058_e0 -15 -15 -10 Ca2+ EX_cpd00063_e0 -15 -100 -10 L-ornithine EX_cpd00064_e0 -0.0068175 -0.00831712 0 H+ EX_cpd00067_e0 -15 -15 -10 L-tyrosine EX_cpd00069_e0 -0.0068175 -0.00233919 0 Sucrose EX_cpd00076_e0 0 -0.02049199 0 L-cysteine EX_cpd00084_e0 -0.0068175 0 0 Cl- EX_cpd00099_e0 -15 -15 -10 Glycerol EX_cpd00100_e0 0 0 -10 Biotin EX_cpd00104_e0 -15 -15 0 D-ribose EX_cpd00105_e0 -0.01862144 0 0 L-leucine EX_cpd00107_e0 -0.03596182 -0.00303228 0 D-galactose EX_cpd00108_e0 -0.25290619 -0.18317325 0 L-histidine EX_cpd00119_e0 -0.0068175 -0.00506825 0 L-proline EX_cpd00129_e0 -0.01102953 0 0 L-malate EX_cpd00130_e0 -0.03649016 -0.79413596 0 D-mannose EX_cpd00138_e0 -0.2540567 -0.05436649 0 Co2 EX_cpd00149_e0 -

Characterization of Aspartylglucosaminidase Activation and Aspartylglucosaminuria Mutations
Arto Pennanen Publications of the National Public Health Institute A 1 / 2004 — INDOOR AIR POLLUTION AND HEALTH RISKS IN FINNISH ICE ARENAS RISKSINFINNISHICE AND HEALTH AIR POLLUTION INDOOR Jani Saarela CHARACTERIZATION OF ASPARTYLGLUCOSAMINIDASE ACTIVATION AND ASPARTYLGLUCOSAMINURIA MUTATIONS ISBN 951-740-485-9 ISSN 0359-3584 ISBN 951-740-486-7 (pdf) Department of Molecular Medicine, ISSN 1458-6290 (pdf) National Public Health Institute, Helsinki, Finland and http://www.ktl.fi /portal/suomi/julkaisut/julkaisusarjat/ Department of Medical Genetics, kansanterveyslaitoksen_julkaisuja_a/ University of Helsinki, Finland Kopijyvä Kuopio 2005 Helsinki 2004 PPennanen_kansi.inddennanen_kansi.indd 1 117.2.20057.2.2005 115:26:195:26:19 CHARACTERIZATION OF ASPARTYLGLUCOSAMINIDASE ACTIVATION AND ASPARTYLGLUCOSAMINURIA MUTATIONS Jani Saarela Department of Molecular Medicine, National Public Health Institute, Helsinki, Finland and Department of Medical Genetics, University of Helsinki, Finland Academic Dissertation To be publicly discussed with the permission of the Medical Faculty of the University of Helsinki, in the lecture room 3 of Biomedicum Helsinki, Haartmaninkatu 8, Helsinki, on January 30th, 2004, at 12 o’clock noon. Helsinki 2004 Supervised by Professor Leena Peltonen-Palotie National Public Health Institute and Department of Medical Genetics University of Helsinki, Helsinki, Finland Reviewed by Professor Ole Kristian Tollersrud and Docent Marc Baumann Department of Medical Biochemistry Protein Chemistry/Proteomics Unit University of Tromsoe and Neuroscience Research Program Tromsoe, Norway University of Helsinki, Helsinki, Finland To be publicly discussed with Professor Marja Makarow Institute of Biotechnology and Department of Applied Biochemistry and Molecular Biology University of Helsinki, Helsinki, Finland Julkaisija-Utgivare-Publisher Kansanterveyslaitos (KTL) Mannerheimintie 166 00300 Helsinki puh. vaihde 09-47441, felefax 09-4744 8408 Folkhälsoinstitutet Mannerheimvägen 166 00300, Helsinki tel.