Lawrence Berkeley National Laboratory Recent Work
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Polarography.Pdf
Polarography UNIT V !1. Polarography Principle The simple principle of polarography is the study of solutions or of electrode processes by means of electrolysis with two electrodes, one polarizable and one unpolarizable, the former formed by mercury regularly dropping from a capillary tube. Polarography is a specific type of measurement that falls into the general category of linear-sweep voltammetry where the electrode potential is altered in a linear fashion from the initial potential to the final potential. As a linear sweep method controlled by convection/diffusion mass transport, the current vs. potential response of a polarographic experiment has the typical sigmoidal shape. What makes polarography different from other linear sweep voltammetry measurements is that polarography makes use of the dropping mercury electrode (DME) or the static mercury drop electrode. Ilkovic Equation Ilkovic equation is a relation used in polarography relating the diffusion current (id) and the concentration of the depolarizer (c), which is the substance reduced or oxidized at the dropping mercury electrode. The Ilkovic equation has the form id = k n D1/3m2/3t1/6c Where k is a constant which includes Faraday constant, π and the density of mercury, and has been evaluated at 708 for max current and 607 for average current, D is the diffusion coefficient of the depolarizer in the medium (cm2/s), n is the number of electrons exchanged in the electrode reaction, m is the mass flow rate of Hg through the capillary (mg/sec), and t is the drop lifetime in seconds, and c is depolarizer concentration in mol/cm3. The equation is named after the scientist who derived it, the Slovak chemist, Dionýz Ilkovič 1907-1980). -

07 Chapter2.Pdf
22 METHODOLOGY 2.1 INTRODUCTION TO ELECTROCHEMICAL TECHNIQUES Electrochemical techniques of analysis involve the measurement of voltage or current. Such methods are concerned with the interplay between solution/electrode interfaces. The methods involve the changes of current, potential and charge as a function of chemical reactions. One or more of the four parameters i.e. potential, current, charge and time can be measured in these techniques and by plotting the graphs of these different parameters in various ways, one can get the desired information. Sensitivity, short analysis time, wide range of temperature, simplicity, use of many solvents are some of the advantages of these methods over the others which makes them useful in kinetic and thermodynamic studies1-3. In general, three electrodes viz., working electrode, the reference electrode, and the counter or auxiliary electrode are used for the measurement in electrochemical techniques. Depending on the combinations of parameters and types of electrodes there are various electrochemical techniques. These include potentiometry, polarography, voltammetry, cyclic voltammetry, chronopotentiometry, linear sweep techniques, amperometry, pulsed techniques etc. These techniques are mainly classified into static and dynamic methods. Static methods are those in which no current passes through the electrode-solution interface and the concentration of analyte species remains constant as in potentiometry. In dynamic methods, a current flows across the electrode-solution interface and the concentration of species changes such as in voltammetry and coulometry4. 2.2 VOLTAMMETRY The field of voltammetry was developed from polarography, which was invented by the Czechoslovakian Chemist Jaroslav Heyrovsky in the early 1920s5. Voltammetry is an electrochemical technique of analysis which includes the measurement of current as a function of applied potential under the conditions that promote polarization of working electrode6. -

Unit 1 Introduction to Electro- Analytical Methods
Introduction to UNIT 1 INTRODUCTION TO ELECTRO- Electroanalytical ANALYTICAL METHODS Methods Structure 1.1 Introduction Objectives 1.2 Basic Concepts Electrical Units Basic Laws of Electrochemistry Electrode Potential Liquid-Junction Potentials Electrochemical Cells The Nernst Equation Cell Potential 1.3 Classification and an Overview of Electroanalytical Methods Potentiometry Voltammetry Polarography Amperometry Electrogravimetry and Coulometry Conductometry 1.4 Classification and Relationships of Electroanalytical Methods 1.5 Summary 1.6 Terminal Questions 1.7 Answers 1.1 INTRODUCTION This is the first unit of this course. This unit deals with the fundamentals of electrochemistry that are necessary for understanding the principles of electroanalytical methods discussed in this Unit 2 to 9. In this unit we have also classified of electroanalytical methods and briefly introduced of some important electroanalytical methods. More details of these elecroanalytical methods will be discussed in the consecutive units. Objectives After studying this unit, you will be able to: • name the different units of electrical quantities, • define the two basic laws of electrochemistry, • describe the single electrode potential and the potential of a galvanic cell, • derive the Nernst expression and give its applications, • calculate the electrode potentials and cell potentials using Nernst equation, • describe the basis for classification of the electroanalytical techniques, and • explain the basis principles and describe the essential conditions of the various electroanalytical techniques. 1.2 BASIC CONCEPTS Before going in detail of different electroanalytical techniques, let’s recapitulate some basic concepts which you have studied in your undergraduate classes. 7 Electroanalytical 1.2.1 Electrical Units Methods -I Ampere (A): Ampere is the unit of current. -

Stationary Electrode Voltammetry and Chronoamperometry in an Alkali Metal Carbonate-Borate Melt
AN ABSTRACT OF THE THESIS OF DARRELL GEORGE PETCOFF for the Doctor of Philosophy (Name of student) (Degree) in Analytical Chemistry presented onC (O,/97 (Major) (Date) Title: STATIONARY ELECTRODE VOLTAMMETRY AND CHRONOAMPEROMETRY IN AN ALKALI METAL CARBONATE - BORATE. MFT T Abstract approved: Redacted for Privacy- Drir. reund The electrochemistry of the lithium-potassium-sodium carbonate-borate melt was explored by voltammetry and chrono- amperometry. In support of this, a controlled-potential polarograph and associated hardware was constructed.Several different types of reference electrodes were tried before choosing a porcelain mem- brane electrode containing a silver wire immersed in a silver sulfate melt.The special porcelain compounded was used also to construct a planar gold disk electrode.The theory of stationary electrode polarography was summarized and denormalized to provide an over- all view. A new approach to the theory of the cyclic background current was also advanced. A computer program was written to facilitate data processing.In addition to providing peak potentials, currents, and n-values, the program also resolves overlapping peaks and furnishes plots of both processed and unprocessed data. Rapid-scan voltammetry was employed to explore the electro- chemical behavior of Zn, Co, Fe, Tl, Sb, As, Ni, Sn, Cd, Te, Bi, Cr, Pb, Cu, and U in the carbonate-borate melt. Most substances gave reasonably well-defined peaks with characteristic peak potentials and n-values.Metal deposition was commonly accompanied by adsorp- tion prepeaks indicative of strong adsorption, and there was also evi- dence of a preceding chemical reaction for several elements, sug- gesting decomplexation before reduction. -

Thesis-1961-B586i.Pdf
INVESTIGATION OF SOME POSSIBILITIES FOR AMPEROMETRIC TITRATION OF CERTAIN METAL IONS WITH OXINE By Donald George Biechler I I Bachelor of Science University of Wisconsin Madison, Wisconsin 1956 Submitted to the faculty of the Graduate School of the Oklahoma State University in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE May, 1961 INVESTIGATION OF SOME POSSIBILITIES FOR AMPEROMEI'RIC TITRATION OF CERTAIN MEI'AL IONS WITH OXINE Thesis Approved: Thesis Adviser i i OKLAHOMA STATE UNIVERSITY llBRARY JAN 2 1962 PREFACE Oxine (8-hydroxyquinoline) is most generally used in analytical chemistry as a precipitant for metals and is known to form water- insoluble chelates with better than thirty metal ions (3). There exists in solutions of oxine a tautomeric equilibrium of the fol- lowing type: C C C C /~/'\ ,c/""/~ C C f ij 1 I II I C C C C C C ~/"'/C N ~/C "+/N r . _I I 0 H 0--------H Chelation of a metal ion involves replacement of the proton and for- mation of a coordinate bond with the nitrogen to form a stable 5 membered ring compound. Thus nickel, a bivalent cation, would form a compound with the following structure: 4810 90 iii iv The oxinates can be ignited and weighed as such or they may be further ignited to the metal oxides and then weighed. Alternately the oxinates may be dissolved in acid and quantitatively brominated (7)0 Considering the number of metal ions that are precipitated by oxine, it seemed that possibly more use could be made of the reagent in volumetric analysis. -
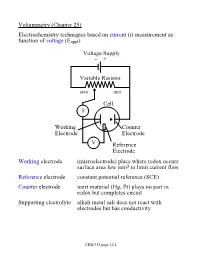
Voltammetry (Chapter 25) Electrochemistry Techniques Based on Current (I) Measurement As Function of Voltage (Eappl)
Voltammetry (Chapter 25) Electrochemistry techniques based on current (i) measurement as function of voltage (Eappl) Voltage Supply - + Variable Resistor max min Cell I Working Counter Electrode Electrode V Reference Electrode Working electrode (microelectrode) place where redox occurs surface area few mm2 to limit current flow Reference electrode constant potential reference (SCE) Counter electrode inert material (Hg, Pt) plays no part in redox but completes circuit Supporting electrolyte alkali metal salt does not react with electrodes but has conductivity CEM 333 page 12.1 Why not use 2 electrodes? OK in potentiometry - very small currents. Now, want to measure current (larger=better) but • potential drops when current is taken from electrode (IR drop) • must minimize current withdrawn from reference electrode surface Potentiostat (voltage source) drives cell • supplies whatever voltage needed between working and counter electrodes to maintain specific voltage between working and reference electrode NOTE: • Almost all current carried between working and counter electrodes • Voltage measured between working and reference electrodes • Analyte dissolved in cell not at electrode surface! CEM 333 page 12.2 Excitation signals (Fig 25-2) CEM 333 page 12.3 Microelectrodes C, Au, Pt, Hg each useful in certain solutions/voltage ranges Fig 25-4 At -ve limit, oxidation of water + - 2H2O ® 4H + O2(g) + 4e At +ve limit, reduction of water - - 2H2O + 2e ® H2 + 2OH CEM 333 page 12.4 Varies with material/solution due to different overpotentials Overpotential -

High Density Carbon Fiber Arrays for Chronic Electrophysiology, Fast Scan Cyclic Voltammetry, and Correlative Anatomy
Journal of Neural Engineering PAPER High density carbon fiber arrays for chronic electrophysiology, fast scan cyclic voltammetry, and correlative anatomy To cite this article: Paras R Patel et al 2020 J. Neural Eng. 17 056029 View the article online for updates and enhancements. This content was downloaded from IP address 141.213.168.11 on 14/10/2020 at 14:43 J. Neural Eng. 17 (2020) 056029 https://doi.org/10.1088/1741-2552/abb1f6 Journal of Neural Engineering PAPER High density carbon fiber arrays for chronic electrophysiology, fast RECEIVED 7 April 2020 scan cyclic voltammetry, and correlative anatomy REVISED 5 August 2020 Paras R Patel1, Pavlo Popov2, Ciara M Caldwell1, Elissa J Welle1, Daniel Egert3, Jeffrey R Pettibone3, ACCEPTED FOR PUBLICATION 4 2,5 3,6 1,7,8,9 4,8,10,11 24 August 2020 Douglas H Roossien , Jill B Becker , Joshua D Berke , Cynthia A Chestek and Dawen Cai 1 PUBLISHED Department of Biomedical Engineering, University of Michigan, Ann Arbor, MI 48109, United States of America 13 October 2020 2 Department of Psychology, University of Michigan, Ann Arbor, MI 48109, United States of America 3 Department of Neurology, University of California, San Francisco, CA 94158, United States of America 4 Department of Cell and Developmental Biology, University of Michigan Medical School, Ann Arbor, MI 48109, United States of America 5 Molecular and Behavioral Neuroscience Institute, University of Michigan, Ann Arbor, MI 48109, United States of America 6 Kavli Institute for Fundamental Neuroscience, University of California, San Francisco, CA 94158, United States of America 7 Department of Electrical Engineering and Computer Science, University of Michigan, Ann Arbor, MI 48109, United States of America 8 Neurosciences Program, University of Michigan, Ann Arbor, MI 48109, United States of America 9 Robotics Program, University of Michigan, Ann Arbor, MI 48109, United States of America 10 Department of Biophysics, University of Michigan, Ann Arbor, MI 48109, United States of America 11 Author to whom any correspondence should be addressed. -

Voltammetric Techniques
Chapter 37 Voltammetric Techniques Samuel P. Kounaves Tufts University Department of Chemistry Summary General Uses • Quantitative determination of organic and inorganic compounds in aqueous and nonaqueous solutions • Measurement of kinetic rates and constants • Determination adsorption processes on surfaces • Determination electron transfer and reaction mechanisms • Determination of thermodynamic properties of solvated species • Fundamental studies of oxidation and reduction processes in various media • Determination of complexation and coordination values Common Applications • Quantitative determination of pharmaceutical compounds • Determination of metal ion concentrations in water to sub–parts-per-billion levels • Determination of redox potentials • Detection of eluted analytes in high-performance liquid chromatography (HPLC) and flow in- jection analysis 709 710 Handbook of Instrumental Techniques for Analytical Chemistry • Determination of number of electrons in redox reactions • Kinetic studies of reactions Samples State Species of interest must be dissolved in an appropriate liquid solvent and capable of being reduced or oxidized within the potential range of the technique and electrode material. Amount The amounts needed to obtain appropriate concentrations vary greatly with the technique. For example, cyclic voltammetry generally requires analyte concentrations of 10–3 to 10–5 M, whereas anodic strip- ping voltammetry of metal ions gives good results with concentrations as low as 10–12 M. Volumes may also vary from about 20 mL to less than a microliter (with special microelectrode cells). Preparation The degree of preparation required depends on both the sample and the technique. For determination of Pb(II) and Cd(II) in seawater with a microelectrode and square-wave anodic stripping voltammetry (ASV), no preparation is required. In contrast, determination of epinepherine in blood plasma at a glassy carbon electrode with differential pulse voltammetry (DPV) requires that the sample first be pre- treated with several reagents, buffered, and separated. -

A Method for the Identification of the Products from Controlled
THACKER, JR., FRANKLIN AUBREY. A Method for the Identification of the Products from Controlled-Potential Coulometry of p-Nitrosophenol, p-Nitrophenol and p-(p-Hydroxyphenylazo)- benzenesulfonic Acid Sodium Salt. (1976) Directed by: Dr. Harvey B. Herman. Pp.44 The aim of this study was to identify the controlled- potential coulometry products after electrolytic reduction of p-nitrosophenol, p-nitrophenol, p-(p-hydroxyphenylazo)- benzenesulfonic acid sodium salt, Orange I, and Orange II. Polarography was performed on all individual compounds in two buffer solutions at pH 4.9 and 11.8 using the dropping mercury electrode polarographic circuit. The resulting polar- ograms were used to select the controlled working electrode potentials for coulometry. Coulometry was conducted with the individual compounds and each pH buffer solution in a stirred mercury pool coulometry cell. The potential was controlled and the number of electrons (n) involved in the reduction process was determined with the aid of an electrical circuit whose output voltage was propor- tional to n. The coulometric reaction solutions were evaporated and the product residues were treated with silylating reagent to form silyl derivatives. The silyl derivatives increased product volatilities so that identification by gas chromatography was possible. Gas chromatography was performed using a packed glass column. The silylated coulometric product was detected by flame ionization. v , As expected, the nitroso, nitro and azo compounds gave coulometric n values equal to 4, 6, and 4 respectively. Also, p-aminophenol was identified as the product after coulometry of p-nitrosophenol, p-nitrophenol, and p-(p-hydroxyphenylazo)- benzenesulfonic acid sodium salt. The coulometry products of Orange I and Orange II were not able to be identified by gas chromatography, apparently because of instability of the anticipated amino-naphthol compounds. -

Polarography
POLAROGRAPHY Electro - analytical technique M. Sc. Paper 4103 – A DEFINATION ! Polarography is a method of analysis in which the solution to be analyzed is electrolyzed under diffusion controlled condition. ! The graph of current (generated) as a function of voltage (applied) is known as POLAROGRAM. The technique is known as POLAROGRAPHY. ! It can be used for qualitative as well as quantitative analysis (inorganic, organic and biological samples) without the requirement of prior separation step (in most of the cases). HISTORY AND BACKGROUND Polarography was created by Jaroslav Heyrovsky in Feb. 10th 1922 1922 1959 On December 10th 1959 he was awarded the Nobel Prize. INTRODUCTION • The basic idea was to pass the current between two electrodes, one having large surface area and other having very small surface area. Both electrodes can be of mercury metal. • The large electrode can be a pool of mercury at the bottom of the cell. • Small electrode is a drop of mercury coming out of a very fine capillary tube, DME. • Thus, if a steady voltage is applied to such a cell, it is possible to construct a reproducible current voltage curve. DROPPING MERCURY ELECTRODE DME Polarography uses regularly renewed mercury drop electrode for analysis INSTRUMENTATION Polarography uses regularly renewed mercury drop electrode for analysis. WORKING ! Electrolyte is a dilute solution of electro active material to be analyzed in a suitable medium containing excess of supporting electrolyte. ! Consider a Polarographic cell, containing a solution of cadmium chloride, to which an external E.M.F is applied. ! The positively charged ions present in the solution will be attracted towards the mercury drop of the dropping mercury electrode (DME). -

Cyclic Voltammetry of Dopamine: an Ec Mechanism
Experiments in Analytical Electrochemistry 4. The Cyclic Voltammetry of Dopamine: an ec mechanism PURPOSE: To determine the kinetic rate of a chemical reaction (c step) that follows an electron- transfer (e step), as illustrated by the oxidation of a neurotransmitter, dopamine. BACKGROUND: Dopamine (DA), 4,5-dihydroxyphenethylamine or 4-(2-aminoethyl)1,2- benzenediol, is a known neurotransmitter that is involved in the chemical transmission of nerve impulses in the mammalian brain. It is a member of the catecholamine family and a precursor to epinephrine (adrenaline) and norepinephrine (noradrenaline) in the biosynthetic pathways. DA has a molecular formula of C8H11NO2 and a formula weight of 153.18 [ref. 1]. It is a water-soluble hormone released by the hypothalamus. Imbalance in dopamine activity can cause brain dysfunction related to two major disorders, Parkinson’s disease and schizophrenia [ref. 2,3]. Researchers are also looking at dopamine neurotransmission in drug abuse ranging from stimulants, such as amphetamines and cocaine, to depressants, such as morphine and other opioids, and alcohol [ref. 3]. Several amine neurotransmitters such as DA, noradrenaline (norepinephrine), adrenaline and serotonin are electroactive so that they can be monitored electrochemically. Most undergo a chemical reaction following the initial electron transfer step, an ec mechanism, as evaluated by cyclic voltammetry (CV) in this experiment. In biological fluids, prior separation with HPLC is recommended in conjunction with an electrochemical detector (HPLC-ECD). Online illustrative applications can be found at http://www.esainc.com/applications/esa_applications.htm. Great strides in learning about the role and fate of DA and other neurotransmitters in brains have come about in recent years due to the ability to monitor these compounds in-vivo. -

Electroanalysis and Coulometric Analysis Allen J
Subscriber access provided by University of Texas Libraries Electroanalysis and Coulometric Analysis Allen J. Bard Anal. Chem., 1966, 38 (5), 88-98• DOI: 10.1021/ac60237a006 • Publication Date (Web): 01 May 2002 Downloaded from http://pubs.acs.org on February 19, 2009 More About This Article The permalink http://dx.doi.org/10.1021/ac60237a006 provides access to: • Links to articles and content related to this article • Copyright permission to reproduce figures and/or text from this article Analytical Chemistry is published by the American Chemical Society. 1155 Sixteenth Street N.W., Washington, DC 20036 fractionating columns are few. Van approach to equilibrium. In most (3) Chem. Eng. Xews 43 (26), June 28, 1965. Sway (17) has developed a fraction packed analytical columns, condenser (4) Elliev, Y. E., Devyatykh, G. G., collector for use in vacuum fractiona- holdup is generally very slight, and Dozorov, 5‘. A., Zh. Fiz. Khim. 37,2179 tion. A piston pump ejects the collected should therefore have only a slight (1963). fractions through a relief valve into effect on rate of equilibration. (5) Ellis, S. R. AI., Porter, XI. C., Jones, K. E., Trans. Znst. Chem. Engrs. (Lon- suitable storage vessels at atmospheric Haring and Knol (7) discuss the don) 41, 212 (1963). pressure. Williams (19) has described influence of reflux ratio on the separating (6) Fair, J. R., Ind. Eng. Chem. 56 (lo), an all-glass fraction cutter with a self- effect in a fractionating column. They October 1964. lubricated valve. This unit is suitable distinguish between separating per- (7).. Haring, H. G., Knol, H.