Transcriptome Sequencing in Pediatric Acute Lymphoblastic Leukemia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Trans Esnps Infers Regulatory Network Architecture
Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2014 © 2014 Anat Kreimer All rights reserved ABSTRACT Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer eSNPs are genetic variants associated with transcript expression levels. The characteristics of such variants highlight their importance and present a unique opportunity for studying gene regulation. eSNPs affect most genes and their cell type specificity can shed light on different processes that are activated in each cell. They can identify functional variants by connecting SNPs that are implicated in disease to a molecular mechanism. Examining eSNPs that are associated with distal genes can provide insights regarding the inference of regulatory networks but also presents challenges due to the high statistical burden of multiple testing. Such association studies allow: simultaneous investigation of many gene expression phenotypes without assuming any prior knowledge and identification of unknown regulators of gene expression while uncovering directionality. This thesis will focus on such distal eSNPs to map regulatory interactions between different loci and expose the architecture of the regulatory network defined by such interactions. We develop novel computational approaches and apply them to genetics-genomics data in human. We go beyond pairwise interactions to define network motifs, including regulatory modules and bi-fan structures, showing them to be prevalent in real data and exposing distinct attributes of such arrangements. We project eSNP associations onto a protein-protein interaction network to expose topological properties of eSNPs and their targets and highlight different modes of distal regulation. -

Clinical Application of Whole Transcriptome Sequencing for The
Walter et al. BMC Cancer (2021) 21:886 https://doi.org/10.1186/s12885-021-08635-5 RESEARCH Open Access Clinical application of whole transcriptome sequencing for the classification of patients with acute lymphoblastic leukemia Wencke Walter1*, Rabia Shahswar2, Anna Stengel1, Manja Meggendorfer1, Wolfgang Kern1, Torsten Haferlach1 and Claudia Haferlach1 Abstract Background: Considering the clinical and genetic characteristics, acute lymphoblastic leukemia (ALL) is a rather heterogeneous hematological neoplasm for which current standard diagnostics require various analyses encompassing morphology, immunophenotyping, cytogenetics, and molecular analysis of gene fusions and mutations. Hence, it would be desirable to rely on a technique and an analytical workflow that allows the simultaneous analysis and identification of all the genetic alterations in a single approach. Moreover, based on the results with standard methods, a significant amount of patients have no established abnormalities and hence, cannot further be stratified. Methods: We performed WTS and WGS in 279 acute lymphoblastic leukemia (ALL) patients (B-cell: n = 211; T-cell: n = 68) to assess the accuracy of WTS, to detect relevant genetic markers, and to classify ALL patients. Results: DNA and RNA-based genotyping was used to ensure correct WTS-WGS pairing. Gene expression analysis reliably assigned samples to the B Cell Precursor (BCP)-ALL or the T-ALL group. Subclassification of BCP-ALL samples was done progressively, assessing first the presence of chromosomal rearrangements by the means of fusion detection. Compared to the standard methods, 97% of the recurrent risk-stratifying fusions could be identified by WTS, assigning 76 samples to their respective entities. Additionally, read-through fusions (indicative of CDKN2A and RB1 gene deletions) were recurrently detected in the cohort along with 57 putative novel fusions, with yet untouched diagnostic potentials. -

Identification of the Binding Partners for Hspb2 and Cryab Reveals
Brigham Young University BYU ScholarsArchive Theses and Dissertations 2013-12-12 Identification of the Binding arP tners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non- Redundant Roles for Small Heat Shock Proteins Kelsey Murphey Langston Brigham Young University - Provo Follow this and additional works at: https://scholarsarchive.byu.edu/etd Part of the Microbiology Commons BYU ScholarsArchive Citation Langston, Kelsey Murphey, "Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins" (2013). Theses and Dissertations. 3822. https://scholarsarchive.byu.edu/etd/3822 This Thesis is brought to you for free and open access by BYU ScholarsArchive. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of BYU ScholarsArchive. For more information, please contact [email protected], [email protected]. Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston A thesis submitted to the faculty of Brigham Young University in partial fulfillment of the requirements for the degree of Master of Science Julianne H. Grose, Chair William R. McCleary Brian Poole Department of Microbiology and Molecular Biology Brigham Young University December 2013 Copyright © 2013 Kelsey Langston All Rights Reserved ABSTRACT Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactors and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston Department of Microbiology and Molecular Biology, BYU Master of Science Small Heat Shock Proteins (sHSP) are molecular chaperones that play protective roles in cell survival and have been shown to possess chaperone activity. -

In Vivo Genome-Wide Binding Interactions of Mouse and Human Constitutive Androstane Receptors Reveal Novel Gene Targets Ben Niu1, Denise M
Published online 8 August 2018 Nucleic Acids Research, 2018, Vol. 46, No. 16 8385–8403 doi: 10.1093/nar/gky692 In vivo genome-wide binding interactions of mouse and human constitutive androstane receptors reveal novel gene targets Ben Niu1, Denise M. Coslo1, Alain R. Bataille2, Istvan Albert2, B. Franklin Pugh2 and Curtis J. Omiecinski1,* 1Center for Molecular Toxicology and Carcinogenesis, Department of Veterinary and Biomedical Sciences, The Pennsylvania State University, University Park, PA 16802, USA and 2Department of Biochemistry and Molecular Biology, The Pennsylvania State University, University Park, PA 16802, USA Received May 04, 2018; Revised July 17, 2018; Editorial Decision July 19, 2018; Accepted July 20, 2018 ABSTRACT tion factors that are activated by direct ligands (1) and gen- erally share a common structure, an N-terminal A/B do- The constitutive androstane receptor (CAR; NR1I3) main, a DNA binding domain (DBD), a hinge domain, a is a nuclear receptor orchestrating complex roles ligand binding/ heterodimerization domain (LBD) and an in cell and systems biology. Species differences in F domain at the C-terminal (2). Upon ligand activation, CAR’s effector pathways remain poorly understood, the LBD would utilize activation function-2 (AF-2) to me- including its role in regulating liver tumor promotion. diate co-regulator interactions (3). However, CAR is dis- We developed transgenic mouse models to assess tinguished from other nuclear receptors in that it lacks an genome-wide binding of mouse and human CAR, A/B domain and is constitutively active in the absence of following receptor activation in liver with direct lig- ligand due to a charge-charge interaction between LBD he- ands and with phenobarbital, an indirect CAR acti- lixes that mimic an active AF-2 conformation, and therefore vator. -

MOZ and MORF, Two Large Mystic Hats in Normal and Cancer Stem Cells
Oncogene (2007) 26, 5408–5419 & 2007 Nature Publishing Group All rights reserved 0950-9232/07 $30.00 www.nature.com/onc REVIEW MOZ and MORF, two large MYSTic HATs in normal and cancer stem cells X-J Yang and M Ullah Molecular Oncology Group, Department of Medicine, McGill University Health Center, Montre´al, Que´bec, Canada Genes of the human monocytic leukemia zinc-finger protein pattern. For cancer biology, it is thus important to MOZ (HUGO symbol, MYST3) and its paralog MORF understand the fundamental mechanisms whereby chro- (MYST4) are rearranged in chromosome translocations matin structure and function are regulated. In the associated withacute myeloid leukemia and/or benign past two decades, our knowledge about regulation in uterine leiomyomata. Both proteins have intrinsic histone this field has exploded. Known regulatory mechanisms acetyltransferase activity and are components of quartet include chromatin assembly, ATP-dependent remodeling, complexes withnoncatalytic subunits containing thebromo- covalent modification, condensin-mediated condensation, domain, plant homeodomain-linked (PHD) finger and replacement with histone variants, and association of proline-tryptophan-tryptophan-proline (PWWP)-containing noncoding RNA (reviewed by Horn and Peterson, 2002; domain, three types of structural modules characteristic of Khorasanizadeh, 2004; Li et al., 2007). Covalent chromatin regulators. Although leukemia-derived fusion pro- modification can occur at both the DNA and histone teins suchas MOZ-TIF2 promote self-renewal of leukemic levels. With histones, modifications include acetylation, stem cells, recent studies indicate that murine MOZ and phosphorylation, methylation, ubiquitination, and many MORF are important for proper development of hema- others (for reviews, see Spencer and Davie, 1999; Strahl topoietic and neurogenic progenitors, respectively, thereby and Allis, 2000; Berger, 2002; Jason et al., 2002; highlighting the importance of epigenetic integrity in Kouzarides, 2007). -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Genome-Wide Profiling of Active Enhancers in Colorectal Cancer
Genome-wide proling of active enhancers in colorectal cancer Min Wu ( [email protected] ) Wuhan University https://orcid.org/0000-0003-1372-4764 Qinglan Li Wuhan University Xiang Lin Wuhan University Ya-Li Yu Zhongnan Hospital, Wuhan University Lin Chen Wuhan University Qi-Xin Hu Wuhan University Meng Chen Zhongnan Hospital, Wuhan University Nan Cao Zhongnan Hospital, Wuhan University Chen Zhao Wuhan University Chen-Yu Wang Wuhan University Cheng-Wei Huang Wuhan University Lian-Yun Li Wuhan University Mei Ye Zhongnan Hospital, Wuhan University https://orcid.org/0000-0002-9393-3680 Article Keywords: Colorectal cancer, H3K27ac, Epigenetics, Enhancer, Transcription factors Posted Date: December 10th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-119156/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Genome-wide profiling of active enhancers in colorectal cancer Qing-Lan Li1, #, Xiang Lin1, #, Ya-Li Yu2, #, Lin Chen1, #, Qi-Xin Hu1, Meng Chen2, Nan Cao2, Chen Zhao1, Chen-Yu Wang1, Cheng-Wei Huang1, Lian-Yun Li1, Mei Ye2,*, Min Wu1,* 1 Frontier Science Center for Immunology and Metabolism, Hubei Key Laboratory of Cell Homeostasis, Hubei Key Laboratory of Developmentally Originated Disease, Hubei Key Laboratory of Intestinal and Colorectal Diseases, College of Life Sciences, Wuhan University, Wuhan, Hubei 430072, China 2Division of Gastroenterology, Department of Geriatrics, Hubei Clinical Centre & Key Laboratory of Intestinal and Colorectal Diseases, Zhongnan Hospital, Wuhan University, Wuhan, Hubei 430072, China #Equal contribution to the study. Contact information *Correspondence should be addressed to Dr. Min Wu, Email: [email protected], Tel: 86-27-68756620, or Dr. -

The Basic Region and Leucine Zipper Transcription Factor Mafk Is a New Nerve Growth Factor-Responsive Immediate Early Gene That Regulates Neurite Outgrowth
The Journal of Neuroscience, October 15, 2002, 22(20):8971–8980 The Basic Region and Leucine Zipper Transcription Factor MafK Is a New Nerve Growth Factor-Responsive Immediate Early Gene That Regulates Neurite Outgrowth Be´ ata To¨ro¨ csik, James M. Angelastro, and Lloyd A. Greene Department of Pathology and Center for Neurobiology and Behavior, Columbia University College of Physicians and Surgeons, New York, New York 10032 We used serial analysis of gene expression to identify new mediated by an atypical isoform of PKC but not by mitogen- NGF-responsive immediate early genes (IEGs) with potential activated kinase kinase, phospholipase C␥, or phosphoinositide roles in neuronal differentiation. Among those identified was 3Ј-kinase. Interference with MafK expression or activity by small MafK, a small Maf family basic region and leucine zipper tran- interfering RNA and dominant negative strategies, respectively, scriptional repressor and coactivator expressed in immature suppresses NGF-promoted outgrowth and maintenance of neu- neurons. NGF treatment elevates the levels of both MafK tran- rites by PC12 cells and neurite outgrowth by immature telence- scripts and protein. In contrast, there is no effect on expression phalic neurons. Our findings support a role for MafK as a novel of the closely related MafG. Unlike many other NGF-responsive regulator of neuronal differentiation. IEGs, MafK regulation shows selectivity and is unresponsive to epidermal growth factor, depolarization, or cAMP derivatives. Key words: MafK; NGF; immediate early -

Aberrant Activity of Histone–Lysine N-Methyltransferase 2 (KMT2) Complexes in Oncogenesis
International Journal of Molecular Sciences Review Aberrant Activity of Histone–Lysine N-Methyltransferase 2 (KMT2) Complexes in Oncogenesis Elzbieta Poreba 1,* , Krzysztof Lesniewicz 2 and Julia Durzynska 1,* 1 Institute of Experimental Biology, Faculty of Biology, Adam Mickiewicz University, ul. Uniwersytetu Pozna´nskiego6, 61-614 Pozna´n,Poland 2 Department of Molecular and Cellular Biology, Institute of Molecular Biology and Biotechnology, Faculty of Biology, Adam Mickiewicz University, ul. Uniwersytetu Pozna´nskiego6, 61-614 Pozna´n,Poland; [email protected] * Correspondence: [email protected] (E.P.); [email protected] (J.D.); Tel.: +48-61-829-5857 (E.P.) Received: 19 November 2020; Accepted: 6 December 2020; Published: 8 December 2020 Abstract: KMT2 (histone-lysine N-methyltransferase subclass 2) complexes methylate lysine 4 on the histone H3 tail at gene promoters and gene enhancers and, thus, control the process of gene transcription. These complexes not only play an essential role in normal development but have also been described as involved in the aberrant growth of tissues. KMT2 mutations resulting from the rearrangements of the KMT2A (MLL1) gene at 11q23 are associated with pediatric mixed-lineage leukemias, and recent studies demonstrate that KMT2 genes are frequently mutated in many types of human cancers. Moreover, other components of the KMT2 complexes have been reported to contribute to oncogenesis. This review summarizes the recent advances in our knowledge of the role of KMT2 complexes in cell transformation. In addition, it discusses the therapeutic targeting of different components of the KMT2 complexes. Keywords: histone–lysine N-methyltransferase 2; COMPASS; COMPASS-like; H3K4 methylation; oncogenesis; cancer; epigenetics; chromatin 1. -

WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization I International Bureau (10) International Publication Number (43) International Publication Date WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT (51) International Patent Classification: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, C12Q 1/68 (2018.01) A61P 31/18 (2006.01) DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, C12Q 1/70 (2006.01) HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, (21) International Application Number: MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, PCT/US2018/056167 OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, (22) International Filing Date: SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, 16 October 2018 (16. 10.2018) TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (26) Publication Language: English GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, (30) Priority Data: UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, 62/573,025 16 October 2017 (16. 10.2017) US TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, FR, GB, GR, HR, HU, ΓΕ , IS, IT, LT, LU, LV, (71) Applicant: MASSACHUSETTS INSTITUTE OF MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, SM, TECHNOLOGY [US/US]; 77 Massachusetts Avenue, TR), OAPI (BF, BJ, CF, CG, CI, CM, GA, GN, GQ, GW, Cambridge, Massachusetts 02139 (US). -
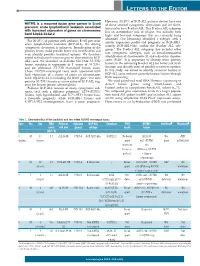
NUTM1 Is a Recurrent Fusion Gene Partner in B-Cell Precursor Acute
LETTERS TO THE EDITOR However, 20-25% of BCP-ALL patients do not have one NUTM1 is a recurrent fusion gene partner in B-cell of these sentinel cytogenetic aberrations and are there- precursor acute lymphoblastic leukemia associated fore said to have B-other ALL. This B-other ALL subgroup with increased expression of genes on chromosome has an intermediate risk of relapse, but includes both band 10p12.31-12.2 high- and low-risk subgroups that are currently being identified. Our laboratory identified a subtype with a For 20-25% of patients with pediatric B-cell precursor similar expression profile and prognosis as BCR-ABL1, acute lymphoblastic leukemia (BCP-ALL), the driving namely BCR-ABL1-like, within the B-other ALL sub- cytogenetic aberration is unknown. Identification of the group.2 The B-other ALL subgroup also includes other primary lesion could provide better risk stratification and rare cytogenetic subtypes, such as intrachromosomal even identify possible treatment options. We therefore amplification of chromosome 21 and a dicentric chromo- aimed to find novel recurrent genetic aberrations in BCP- 1 ALL cases. We identified an in-frame SLC12A6-NUTM1 some (9;20). It is important to identify more primary fusion, resulting in expression of 3’ exons of NUTM1, lesions in the remaining B-other ALL for better risk strat- and six additional NUTM1-rearranged fusion cases. ification and identification of possible treatment options. These NUTM1-rearranged cases were associated with In this study, we aimed to identify recurrent fusions in high expression of a cluster of genes on chromosome BCP-ALL cases without currently known lesions through band 10p12.31-12.2, including the BMI1 gene. -

Mutational Landscape and Clinical Outcome of Patients with De Novo Acute Myeloid Leukemia and Rearrangements Involving 11Q23/KMT2A
Mutational landscape and clinical outcome of patients with de novo acute myeloid leukemia and rearrangements involving 11q23/KMT2A Marius Billa,1,2, Krzysztof Mrózeka,1,2, Jessica Kohlschmidta,b, Ann-Kathrin Eisfelda,c, Christopher J. Walkera, Deedra Nicoleta,b, Dimitrios Papaioannoua, James S. Blachlya,c, Shelley Orwicka,c, Andrew J. Carrolld, Jonathan E. Kolitze, Bayard L. Powellf, Richard M. Stoneg, Albert de la Chapelleh,i,2, John C. Byrda,c, and Clara D. Bloomfielda,c aThe Ohio State University Comprehensive Cancer Center, Columbus, OH 43210; bAlliance for Clinical Trials in Oncology Statistics and Data Center, The Ohio State University Comprehensive Cancer Center, Columbus, OH 43210; cDivision of Hematology, Department of Internal Medicine, The Ohio State University Comprehensive Cancer Center, Columbus, OH 43210; dDepartment of Genetics, University of Alabama at Birmingham, Birmingham, AL 35294; eNorthwell Health Cancer Institute, Zucker School of Medicine at Hofstra/Northwell, Lake Success, NY 11042; fDepartment of Internal Medicine, Section on Hematology & Oncology, Wake Forest Baptist Comprehensive Cancer Center, Winston-Salem, NC 27157; gDepartment of Medical Oncology, Dana-Farber/Partners Cancer Care, Boston, MA 02215; hHuman Cancer Genetics Program, Comprehensive Cancer Center, The Ohio State University, Columbus, OH 43210; and iDepartment of Cancer Biology and Genetics, Comprehensive Cancer Center, The Ohio State University, Columbus, OH 43210 Contributed by Albert de la Chapelle, August 28, 2020 (sent for review July 17, 2020; reviewed by Anne Hagemeijer and Stefan Klaus Bohlander) Balanced rearrangements involving the KMT2A gene, located at patterns that include high expression of HOXA genes and thereby 11q23, are among the most frequent chromosome aberrations in contribute to leukemogenesis (14–16).