Age=F(Physical Activity)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

University of California, San Diego
UNIVERSITY OF CALIFORNIA, SAN DIEGO The post-terminal differentiation fate of RNAs revealed by next-generation sequencing A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Biomedical Sciences by Gloria Kuo Lefkowitz Committee in Charge: Professor Benjamin D. Yu, Chair Professor Richard Gallo Professor Bruce A. Hamilton Professor Miles F. Wilkinson Professor Eugene Yeo 2012 Copyright Gloria Kuo Lefkowitz, 2012 All rights reserved. The Dissertation of Gloria Kuo Lefkowitz is approved, and it is acceptable in quality and form for publication on microfilm and electronically: __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ Chair University of California, San Diego 2012 iii DEDICATION Ma and Ba, for your early indulgence and support. Matt and James, for choosing more practical callings. Roy, my love, for patiently sharing the ups and downs of this journey. iv EPIGRAPH It is foolish to tear one's hair in grief, as though sorrow would be made less by baldness. ~Cicero v TABLE OF CONTENTS Signature Page .............................................................................................................. iii Dedication .................................................................................................................... -

Rap1-Mediated Chromatin and Gene Expression Changes at Senescence
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2019 Rap1-Mediated Chromatin And Gene Expression Changes At Senescence Shufei Song University of Pennsylvania Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Biochemistry Commons, and the Cell Biology Commons Recommended Citation Song, Shufei, "Rap1-Mediated Chromatin And Gene Expression Changes At Senescence" (2019). Publicly Accessible Penn Dissertations. 3557. https://repository.upenn.edu/edissertations/3557 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/3557 For more information, please contact [email protected]. Rap1-Mediated Chromatin And Gene Expression Changes At Senescence Abstract ABSTRACT RAP1-MEDIATED CHROMATIN AND GENE EXPRESSION CHANGES AT SENESCENCE The telomeric protein Rap1 has been extensively studied for its roles as a transcriptional activator and repressor. Indeed, in both yeast and mammals, Rap1 is known to bind throughout the genome to reorganize chromatin and regulate gene transcription. Previously, our lab published evidence that Rap1 plays important roles in cellular senescence. In telomerase-deficient S. cerevisiae, Rap1 relocalizes from telomeres and subtelomeres to new Rap1 target at senescence (NRTS). This leads to two types of histone loss: Rap1 lowers global histone levels by repressing histone gene transcription and it also results in local nucleosome displacement at the promoters of the activated NRTS. Here, I examine mechanisms of site-specific histone loss by presenting evidence that Rap1 can directly interact with histone tetramers H3/H4, and map this interaction to a three-amino-acid-patch within the DNA binding domain. Functional studies are performed in vivo using a mutant form of Rap1 with weakened histone interactions, and deficient promoter clearance as well as blunted gene activation is observed, indicating that direct Rap1-H3/H4 interactions are involved in nucleosome displacement. -

Multi-Trait Analysis for Genome-Wide Association Study of Five Psychiatric
Wu et al. Translational Psychiatry (2020) 10:209 https://doi.org/10.1038/s41398-020-00902-6 Translational Psychiatry ARTICLE Open Access Multi-trait analysis for genome-wide association study of five psychiatric disorders Yulu Wu1,2, Hongbao Cao3, Ancha Baranova4,5, Hailiang Huang6,7,ShengLi8,LeiCai8,ShuquanRao9, Minhan Dai1,2, Min Xie1,2,YikaiDou1,2, Qinjian Hao 10,LingZhu1,2, Xiangrong Zhang11,YinYao12, Fuquan Zhang 13, Mingqing Xu8,14 and Qiang Wang 1,2 Abstract We conducted a cross-trait meta-analysis of genome-wide association study on schizophrenia (SCZ) (n = 65,967), bipolar disorder (BD) (n = 41,653), autism spectrum disorder (ASD) (n = 46,350), attention deficit hyperactivity disorder (ADHD) (n = 55,374), and depression (DEP) (n = 688,809). After the meta-analysis, the number of genomic loci increased from 14 to 19 in ADHD, from 3 to 10 in ASD, from 45 to 57 in DEP, from 8 to 54 in BD, and from 64 to 87 in SCZ. We observed significant enrichment of overlapping genes among different disorders and identified a panel of cross-disorder genes. A total of seven genes were found being commonly associated with four out of five psychiatric conditions, namely GABBR1, GLT8D1, HIST1H1B, HIST1H2BN, HIST1H4L, KCNB1, and DCC. The SORCS3 gene was highlighted due to the fact that it was involved in all the five conditions of study. Analysis of correlations unveiled the existence of two clusters of related psychiatric conditions, SCZ and BD that were separate from the other three traits, and formed another group. Our results may provide a new insight for genetic basis of the five psychiatric disorders. -

Identification of Key Genes and Pathways for Alzheimer's Disease
Biophys Rep 2019, 5(2):98–109 https://doi.org/10.1007/s41048-019-0086-2 Biophysics Reports RESEARCH ARTICLE Identification of key genes and pathways for Alzheimer’s disease via combined analysis of genome-wide expression profiling in the hippocampus Mengsi Wu1,2, Kechi Fang1, Weixiao Wang1,2, Wei Lin1,2, Liyuan Guo1,2&, Jing Wang1,2& 1 CAS Key Laboratory of Mental Health, Institute of Psychology, Chinese Academy of Sciences, Beijing 100101, China 2 Department of Psychology, University of Chinese Academy of Sciences, Beijing 10049, China Received: 8 August 2018 / Accepted: 17 January 2019 / Published online: 20 April 2019 Abstract In this study, combined analysis of expression profiling in the hippocampus of 76 patients with Alz- heimer’s disease (AD) and 40 healthy controls was performed. The effects of covariates (including age, gender, postmortem interval, and batch effect) were controlled, and differentially expressed genes (DEGs) were identified using a linear mixed-effects model. To explore the biological processes, func- tional pathway enrichment and protein–protein interaction (PPI) network analyses were performed on the DEGs. The extended genes with PPI to the DEGs were obtained. Finally, the DEGs and the extended genes were ranked using the convergent functional genomics method. Eighty DEGs with q \ 0.1, including 67 downregulated and 13 upregulated genes, were identified. In the pathway enrichment analysis, the 80 DEGs were significantly enriched in one Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway, GABAergic synapses, and 22 Gene Ontology terms. These genes were mainly involved in neuron, synaptic signaling and transmission, and vesicle metabolism. These processes are all linked to the pathological features of AD, demonstrating that the GABAergic system, neurons, and synaptic function might be affected in AD. -

University of California, San Diego
UC San Diego UC San Diego Electronic Theses and Dissertations Title The post-terminal differentiation fate of RNAs revealed by next-generation sequencing Permalink https://escholarship.org/uc/item/7324r1rj Author Lefkowitz, Gloria Kuo Publication Date 2012 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA, SAN DIEGO The post-terminal differentiation fate of RNAs revealed by next-generation sequencing A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Biomedical Sciences by Gloria Kuo Lefkowitz Committee in Charge: Professor Benjamin D. Yu, Chair Professor Richard Gallo Professor Bruce A. Hamilton Professor Miles F. Wilkinson Professor Eugene Yeo 2012 Copyright Gloria Kuo Lefkowitz, 2012 All rights reserved. The Dissertation of Gloria Kuo Lefkowitz is approved, and it is acceptable in quality and form for publication on microfilm and electronically: __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ Chair University of California, San Diego 2012 iii DEDICATION Ma and Ba, for your early indulgence and support. Matt and James, for choosing more practical callings. Roy, my love, for patiently sharing the ups and downs -

The Human Canonical Core Histone Catalogue David Miguel Susano Pinto*, Andrew Flaus*,†
bioRxiv preprint doi: https://doi.org/10.1101/720235; this version posted July 30, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. The Human Canonical Core Histone Catalogue David Miguel Susano Pinto*, Andrew Flaus*,† Abstract Core histone proteins H2A, H2B, H3, and H4 are encoded by a large family of genes dis- tributed across the human genome. Canonical core histones contribute the majority of proteins to bulk chromatin packaging, and are encoded in 4 clusters by 65 coding genes comprising 17 for H2A, 18 for H2B, 15 for H3, and 15 for H4, along with at least 17 total pseudogenes. The canonical core histone genes display coding variation that gives rise to 11 H2A, 15 H2B, 4 H3, and 2 H4 unique protein isoforms. Although histone proteins are highly conserved overall, these isoforms represent a surprising and seldom recognised variation with amino acid identity as low as 77 % between canonical histone proteins of the same type. The gene sequence and protein isoform diversity also exceeds com- monly used subtype designations such as H2A.1 and H3.1, and exists in parallel with the well-known specialisation of variant histone proteins. RNA sequencing of histone transcripts shows evidence for differential expression of histone genes but the functional significance of this variation has not yet been investigated. To assist understanding of the implications of histone gene and protein diversity we have catalogued the entire human canonical core histone gene and protein complement. -

A Multiprotein Occupancy Map of the Mrnp on the 3 End of Histone
Downloaded from rnajournal.cshlp.org on October 6, 2021 - Published by Cold Spring Harbor Laboratory Press A multiprotein occupancy map of the mRNP on the 3′ end of histone mRNAs LIONEL BROOKS III,1 SHAWN M. LYONS,2 J. MATTHEW MAHONEY,1 JOSHUA D. WELCH,3 ZHONGLE LIU,1 WILLIAM F. MARZLUFF,2 and MICHAEL L. WHITFIELD1 1Department of Genetics, Dartmouth Geisel School of Medicine, Hanover, New Hampshire 03755, USA 2Integrative Program for Biological and Genome Sciences, University of North Carolina, Chapel Hill, North Carolina 27599, USA 3Department of Computer Science, University of North Carolina, Chapel Hill, North Carolina 27599, USA ABSTRACT The animal replication-dependent (RD) histone mRNAs are coordinately regulated with chromosome replication. The RD-histone mRNAs are the only known cellular mRNAs that are not polyadenylated. Instead, the mature transcripts end in a conserved stem– loop (SL) structure. This SL structure interacts with the stem–loop binding protein (SLBP), which is involved in all aspects of RD- histone mRNA metabolism. We used several genomic methods, including high-throughput sequencing of cross-linked immunoprecipitate (HITS-CLIP) to analyze the RNA-binding landscape of SLBP. SLBP was not bound to any RNAs other than histone mRNAs. We performed bioinformatic analyses of the HITS-CLIP data that included (i) clustering genes by sequencing read coverage using CVCA, (ii) mapping the bound RNA fragment termini, and (iii) mapping cross-linking induced mutation sites (CIMS) using CLIP-PyL software. These analyses allowed us to identify specific sites of molecular contact between SLBP and its RD-histone mRNA ligands. We performed in vitro crosslinking assays to refine the CIMS mapping and found that uracils one and three in the loop of the histone mRNA SL preferentially crosslink to SLBP, whereas uracil two in the loop preferentially crosslinks to a separate component, likely the 3′hExo. -

Acetyl-Histone H4-K12 Pab Cat
Acetyl-Histone H4-K12 pAb Cat. No.: 15-437 Acetyl-Histone H4-K12 pAb Immunohistochemistry of paraffin- embedded rat ovary using Acetyl-Histone Immunohistochemistry of paraffin-embedded mouse testis using H4-K12 antibody (15-437) at dilution of 1:200 Acetyl-Histone H4-K12 antibody (15-437) at dilution of 1:200 (40x lens). (40x lens). Immunohistochemistry of paraffin- Immunofluorescence analysis of C6 cells using Acetyl-Histone H4-K12 embedded mouse spinal cord using Acetyl- antibody (15-437) at dilution of 1:100.C6 cells were treated by TSA (1 Histone H4-K12 antibody (15-437) at dilution uM) at 37? for 18 hours. Blue: DAPI for nuclear staining. of 1:200 (40x lens). September 27, 2021 1 https://www.prosci-inc.com/acetyl-histone-h4-k12-pab-15-437.html Immunofluorescence analysis of NIH/3T3 cells using Acetyl-Histone H4-K12 antibody (15-437) at dilution of 1:100.NIH/3T3 cells were treated by TSA (1 uM) at 37? for 18 hours. Blue: DAPI for nuclear staining. Specifications HOST SPECIES: Rabbit SPECIES REACTIVITY: Human, Mouse IMMUNOGEN: A synthetic acetylated peptide around K12 of human Histone H4 (NP_001029249.1). TESTED APPLICATIONS: IF, IHC, WB WB: ,1:500 - 1:2000 APPLICATIONS: IHC: ,1:50 - 1:200 IF: ,1:50 - 1:200 Properties PURIFICATION: Affinity purification CLONALITY: Polyclonal ISOTYPE: IgG CONJUGATE: Unconjugated PHYSICAL STATE: Liquid BUFFER: PBS with 0.02% sodium azide, 50% glycerol, pH7.3. STORAGE CONDITIONS: Store at -20˚C. Avoid freeze / thaw cycles. Additional Info OFFICIAL SYMBOL: H4K12ac September 27, 2021 2 https://www.prosci-inc.com/acetyl-histone-h4-k12-pab-15-437.html H4/A, H4FA, HIST1H4B, H4/I, H4FI, HIST1H4C, H4/G, H4FG, HIST1H4D, H4/B, H4FB, HIST1H4E, H4/J, H4FJ, HIST1H4F, H4/C, H4FC, HIST1H4H, H4/H, H4FH, HIST1H4I, H4/M, ALTERNATE NAMES: H4FM, HIST1H4J, H4/E, H4FE, HIST1H4K, H4/D, H4FD, HIST1H4L, H4/K, H4FK, HIST2H4A, H4/N, H4F2, H4FN, HIST2H4, HIST2H4B, H4/O, H4FO, HIST4H4, Histone H4H4FA GENE ID: 8366 USER NOTE: Optimal dilutions for each application to be determined by the researcher. -
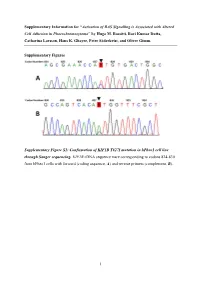
Activation of RAS Signalling Is Associated with Altered Cell Adhesion in Phaeochromocytoma” by Hugo M
Supplementary Information for “Activation of RAS Signalling is Associated with Altered Cell Adhesion in Phaeochromocytoma” by Hugo M. Rossitti, Ravi Kumar Dutta, Catharina Larsson, Hans K. Ghayee, Peter Söderkvist, and Oliver Gimm. Supplementary Figures Supplementary Figure S1: Confirmation of KIF1B T827I mutation in hPheo1 cell line through Sanger sequencing. KIF1B cDNA sequence trace corresponding to codons 824-830 from hPheo1 cells with forward (coding sequence, A) and reverse primers (complement, B). 1 Supplementary Figure S2: Confirmation of NRAS Q61K mutation in hPheo1 cell line through Sanger sequencing. NRAS cDNA sequence trace corresponding to codons 56-67 from hPheo1 cells with forward (coding sequence, A) and reverse primers (complement, B). 2 Supplementary Figure S3: CCND1 gene expression and hPheo1 proliferation. A: Expression of CCND1 mRNA assessed by RT-qPCR and presented as fold change (2-ΔΔCT, mean ± standard error of the mean). B: Cell counts at 1, 2, and 3 days after plating (corresponding to 72, 96 and 120 hours posttransfection, respectively) of control- or siNRAS#1-transfected hPheo1 cells expressed as fold change of the number of cells plated at day 0 (48 hours posttransfection; mean ± standard deviation). All results are from three independent siRNA experiments. 3 Supplementary Tables Supplementary Table S1: List of transcript cluster IDs significantly upregulated in hPheo1 by siNRAS treatment (comparison: siNRAS versus control-transfected hPheo1; ANOVA p < 0.05, FDR < 0.25, fold change < -1.5 or > 1.5). Transcript -

( 12 ) Patent Application Publication ( 10 ) Pub . No .: US 2021/0005284 A1 Nuzhdina Et Al
US 20210005284A1 IN ( 19 ) United States ( 12 ) Patent Application Publication ( 10 ) Pub . No .: US 2021/0005284 A1 Nuzhdina et al . ( 43 ) Pub . Date : Jan. 7 , 2021 ( 54 ) TECHNIQUES FOR NUCLEIC ACID DATA G16B 25/10 ( 2006.01 ) QUALITY CONTROL G16B 35/10 ( 2006.01 ) ( 52 ) U.S. CI . ( 71 ) Applicant: BostonGene Corporation , Waltham , CPC G16B 30/10 ( 2019.02 ) ; G16B 35/10 MA ( US ) ( 2019.02 ) ; G16B 25/10 ( 2019.02 ) ; G16B ( 72 ) Inventors : Ekaterina Nuzhdina , Moscow ( RU ); 20/20 ( 2019.02 ) Alexander Bagaev , Moscow ( RU ); Maksim Chelushkin , Moscow (RU ) ; Yaroslav Lozinsky, Moscow ( RU ); ( 57 ) ABSTRACT Natalia Miheecheva , Moscow ( RU ) ; Alexander Zaitsev , Leninskiy R - N (RU ) Described herein are various methods of collecting and processing of tumor and /or healthy tissue samples to extract ( 21 ) Appl . No .: 16 / 920,641 nucleic acid and perform nucleic acid sequencing . Also described herein are various methods of processing nucleic ( 22 ) Filed : Jul . 3 , 2020 acid sequencing data to remove bias from the nucleic acid sequencing data . Also described herein are various methods Related U.S. Application Data of evaluating the quality of nucleic acid sequence informa ( 60 ) Provisional application No. 62 / 991,570 , filed on Mar. tion . The identity and / or integrity of nucleic acid sequence 18 , 2020 , provisional application No. 62 /870,622 , data is evaluated prior to using the sequence information for filed on Jul. 3 , 2019 . subsequent analysis ( for example for diagnostic , prognostic , or clinical purposes ). The methods enable a subject, doctor, Publication Classification or user to characterize or classify various types of cancer ( 51 ) Int . Cl . precisely , and thereby determine a therapy or combination of G16B 30/10 ( 2006.01 ) therapies that may be effective to treat a cancer in a subject G16B 20/20 ( 2006.01 ) based on the precise characterization . -

The Human Histone H3 Complement Anno 2011
Biochimica et Biophysica Acta 1809 (2011) 577–586 Contents lists available at ScienceDirect Biochimica et Biophysica Acta journal homepage: www.elsevier.com/locate/bbagrm Review The human histone H3 complement anno 2011 Thomas H.A. Ederveen, Imke K. Mandemaker, Colin Logie ⁎ Department of Molecular Biology, Nijmegen Centre for Molecular Life Sciences, Radboud University, The Netherlands article info abstract Article history: Histones are highly basic, relatively small proteins that complex with DNA to form higher order structures Received 31 May 2011 that underlie chromosome topology. Of the four core histones H2A, H2B, H3 and H4, it is H3 that is most Received in revised form 5 July 2011 heavily modified at the post-translational level. The human genome harbours 16 annotated bona fide histone Accepted 6 July 2011 H3 genes which code for four H3 protein variants. In 2010, two novel histone H3.3 protein variants were Available online 14 July 2011 reported, carrying over twenty amino acid substitutions. Nevertheless, they appear to be incorporated into chromatin. Interestingly, these new H3 genes are located on human chromosome 5 in a repetitive region that Keywords: fi fi Human histone H3 harbours an additional ve H3 pseudogenes, but no other core histone ORFs. In addition, a human-speci c Protein variant novel putative histone H3.3 variant located at 12p11.21 was reported in 2011. These developments raised the Pseudogene question as to how many more human histone H3 ORFs there may be. Using homology searches, we detected 41 histone H3 pseudogenes in the current human genome assembly. The large majority are derived from the H3.3 gene H3F3A, and three of those may code for yet more histone H3.3 protein variants. -

Downloaded from 160 the UCSC Genome Bioinformatics Site
bioRxiv preprint doi: https://doi.org/10.1101/069351; this version posted August 12, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Recurrent promoter mutations in melanoma are defined by an extended 2 context-specific mutational signature 3 Johan Fredriksson1 and Erik Larsson1* 4 1Department of Medical Biochemistry and Cell Biology, Institute of Biomedicine, The 5 Sahlgrenska Academy, University of Gothenburg, SE-405 30 Gothenburg, Sweden. 6 *Correspondence to [email protected] 7 Summary 8 Sequencing of whole tumor genomes holds the promise of revealing functional somatic 9 regulatory mutations, such as those described in the TERT promoter1,2. Recurrent 10 promoter mutations have been identified in many additional genes3,4 and appear to be 11 particularly common in melanoma5,6, but convincing functional data such influence on 12 gene expression has been elusive. Here, we show that frequently recurring promoter 13 mutations in melanoma occur almost exclusively at cytosines flanked by an extended 14 non-degenerate sequence signature, TTCCG, with TERT as a notable exception. In 15 active, but not inactive, promoters, mutation frequencies for cytosines at the 5’ end of 16 this ETS-like motif were considerably higher than expected based on a UV trinucleotide 17 mutation signature. Additional analyses solidify this pattern as an extended context- 18 specific mutational signature that mediates an exceptional position-specific elevation in 19 local mutation rate, arguing against positive selection. This finding has implications for 20 the interpretation of somatic mutations in regulatory regions, and underscores the 21 importance of genomic context and extended sequence patterns to accurately describe 22 mutational signatures in cancer.