Downloaded from 160 the UCSC Genome Bioinformatics Site
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Old Data and Friends Improve with Age: Advancements with the Updated Tools of Genenetwork
bioRxiv preprint doi: https://doi.org/10.1101/2021.05.24.445383; this version posted May 25, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Old data and friends improve with age: Advancements with the updated tools of GeneNetwork Alisha Chunduri1, David G. Ashbrook2 1Department of Biotechnology, Chaitanya Bharathi Institute of Technology, Hyderabad 500075, India 2Department of Genetics, Genomics and Informatics, University of Tennessee Health Science Center, Memphis, TN 38163, USA Abstract Understanding gene-by-environment interactions is important across biology, particularly behaviour. Families of isogenic strains are excellently placed, as the same genome can be tested in multiple environments. The BXD’s recent expansion to 140 strains makes them the largest family of murine isogenic genomes, and therefore give great power to detect QTL. Indefinite reproducible genometypes can be leveraged; old data can be reanalysed with emerging tools to produce novel biological insights. To highlight the importance of reanalyses, we obtained drug- and behavioural-phenotypes from Philip et al. 2010, and reanalysed their data with new genotypes from sequencing, and new models (GEMMA and R/qtl2). We discover QTL on chromosomes 3, 5, 9, 11, and 14, not found in the original study. We narrowed down the candidate genes based on their ability to alter gene expression and/or protein function, using cis-eQTL analysis, and variants predicted to be deleterious. Co-expression analysis (‘gene friends’) and human PheWAS were used to further narrow candidates. -

University of California, San Diego
UNIVERSITY OF CALIFORNIA, SAN DIEGO The post-terminal differentiation fate of RNAs revealed by next-generation sequencing A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Biomedical Sciences by Gloria Kuo Lefkowitz Committee in Charge: Professor Benjamin D. Yu, Chair Professor Richard Gallo Professor Bruce A. Hamilton Professor Miles F. Wilkinson Professor Eugene Yeo 2012 Copyright Gloria Kuo Lefkowitz, 2012 All rights reserved. The Dissertation of Gloria Kuo Lefkowitz is approved, and it is acceptable in quality and form for publication on microfilm and electronically: __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ Chair University of California, San Diego 2012 iii DEDICATION Ma and Ba, for your early indulgence and support. Matt and James, for choosing more practical callings. Roy, my love, for patiently sharing the ups and downs of this journey. iv EPIGRAPH It is foolish to tear one's hair in grief, as though sorrow would be made less by baldness. ~Cicero v TABLE OF CONTENTS Signature Page .............................................................................................................. iii Dedication .................................................................................................................... -

The Basal Bodies of Chlamydomonas Reinhardtii Susan K
Dutcher and O’Toole Cilia (2016) 5:18 DOI 10.1186/s13630-016-0039-z Cilia REVIEW Open Access The basal bodies of Chlamydomonas reinhardtii Susan K. Dutcher1* and Eileen T. O’Toole2 Abstract The unicellular green alga, Chlamydomonas reinhardtii, is a biflagellated cell that can swim or glide. C. reinhardtii cells are amenable to genetic, biochemical, proteomic, and microscopic analysis of its basal bodies. The basal bodies contain triplet microtubules and a well-ordered transition zone. Both the mother and daughter basal bodies assemble flagella. Many of the proteins found in other basal body-containing organisms are present in the Chlamydomonas genome, and mutants in these genes affect the assembly of basal bodies. Electron microscopic analysis shows that basal body duplication is site-specific and this may be important for the proper duplication and spatial organization of these organelles. Chlamydomonas is an excellent model for the study of basal bodies as well as the transition zone. Keywords: Site-specific basal body duplication, Cartwheel, Transition zone, Centrin fibers Phylogeny and conservation of proteins Centrin, SPD2/CEP192, Asterless/CEP152; CEP70, The green lineage or Viridiplantae consists of the green delta-tubulin, and epsilon-tubulin. Chlamydomonas has algae, which include Chlamydomonas, the angiosperms homologs of all of these based on sequence conservation (the land plants), and the gymnosperms (conifers, cycads, except PLK4, CEP152, and CEP192. Several lines of evi- ginkgos). They are grouped together because they have dence suggests that CEP152, CEP192, and PLK4 interact chlorophyll a and b and lack phycobiliproteins. The green [20, 52] and their concomitant absence in several organ- algae together with the cycads and ginkgos have basal isms suggests that other mechanisms exist that allow for bodies and cilia, while the angiosperms and conifers have control of duplication [4]. -

Par6c Is at the Mother Centriole and Controls Centrosomal Protein
860 Research Article Par6c is at the mother centriole and controls centrosomal protein composition through a Par6a-dependent pathway Vale´rian Dormoy, Kati Tormanen and Christine Su¨ tterlin* Department of Developmental and Cell Biology, University of California, Irvine, Irvine, CA 92697-2300, USA *Author for correspondence ([email protected]) Accepted 3 December 2012 Journal of Cell Science 126, 860–870 ß 2013. Published by The Company of Biologists Ltd doi: 10.1242/jcs.121186 Summary The centrosome contains two centrioles that differ in age, protein composition and function. This non-membrane bound organelle is known to regulate microtubule organization in dividing cells and ciliogenesis in quiescent cells. These specific roles depend on protein appendages at the older, or mother, centriole. In this study, we identified the polarity protein partitioning defective 6 homolog gamma (Par6c) as a novel component of the mother centriole. This specific localization required the Par6c C-terminus, but was independent of intact microtubules, the dynein/dynactin complex and the components of the PAR polarity complex. Par6c depletion resulted in altered centrosomal protein composition, with the loss of a large number of proteins, including Par6a and p150Glued, from the centrosome. As a consequence, there were defects in ciliogenesis, microtubule organization and centrosome reorientation during migration. Par6c interacted with Par3 and aPKC, but these proteins were not required for the regulation of centrosomal protein composition. Par6c also associated with Par6a, which controls protein recruitment to the centrosome through p150Glued. Our study is the first to identify Par6c as a component of the mother centriole and to report a role of a mother centriole protein in the regulation of centrosomal protein composition. -

Rap1-Mediated Chromatin and Gene Expression Changes at Senescence
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2019 Rap1-Mediated Chromatin And Gene Expression Changes At Senescence Shufei Song University of Pennsylvania Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Biochemistry Commons, and the Cell Biology Commons Recommended Citation Song, Shufei, "Rap1-Mediated Chromatin And Gene Expression Changes At Senescence" (2019). Publicly Accessible Penn Dissertations. 3557. https://repository.upenn.edu/edissertations/3557 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/3557 For more information, please contact [email protected]. Rap1-Mediated Chromatin And Gene Expression Changes At Senescence Abstract ABSTRACT RAP1-MEDIATED CHROMATIN AND GENE EXPRESSION CHANGES AT SENESCENCE The telomeric protein Rap1 has been extensively studied for its roles as a transcriptional activator and repressor. Indeed, in both yeast and mammals, Rap1 is known to bind throughout the genome to reorganize chromatin and regulate gene transcription. Previously, our lab published evidence that Rap1 plays important roles in cellular senescence. In telomerase-deficient S. cerevisiae, Rap1 relocalizes from telomeres and subtelomeres to new Rap1 target at senescence (NRTS). This leads to two types of histone loss: Rap1 lowers global histone levels by repressing histone gene transcription and it also results in local nucleosome displacement at the promoters of the activated NRTS. Here, I examine mechanisms of site-specific histone loss by presenting evidence that Rap1 can directly interact with histone tetramers H3/H4, and map this interaction to a three-amino-acid-patch within the DNA binding domain. Functional studies are performed in vivo using a mutant form of Rap1 with weakened histone interactions, and deficient promoter clearance as well as blunted gene activation is observed, indicating that direct Rap1-H3/H4 interactions are involved in nucleosome displacement. -

Multi-Trait Analysis for Genome-Wide Association Study of Five Psychiatric
Wu et al. Translational Psychiatry (2020) 10:209 https://doi.org/10.1038/s41398-020-00902-6 Translational Psychiatry ARTICLE Open Access Multi-trait analysis for genome-wide association study of five psychiatric disorders Yulu Wu1,2, Hongbao Cao3, Ancha Baranova4,5, Hailiang Huang6,7,ShengLi8,LeiCai8,ShuquanRao9, Minhan Dai1,2, Min Xie1,2,YikaiDou1,2, Qinjian Hao 10,LingZhu1,2, Xiangrong Zhang11,YinYao12, Fuquan Zhang 13, Mingqing Xu8,14 and Qiang Wang 1,2 Abstract We conducted a cross-trait meta-analysis of genome-wide association study on schizophrenia (SCZ) (n = 65,967), bipolar disorder (BD) (n = 41,653), autism spectrum disorder (ASD) (n = 46,350), attention deficit hyperactivity disorder (ADHD) (n = 55,374), and depression (DEP) (n = 688,809). After the meta-analysis, the number of genomic loci increased from 14 to 19 in ADHD, from 3 to 10 in ASD, from 45 to 57 in DEP, from 8 to 54 in BD, and from 64 to 87 in SCZ. We observed significant enrichment of overlapping genes among different disorders and identified a panel of cross-disorder genes. A total of seven genes were found being commonly associated with four out of five psychiatric conditions, namely GABBR1, GLT8D1, HIST1H1B, HIST1H2BN, HIST1H4L, KCNB1, and DCC. The SORCS3 gene was highlighted due to the fact that it was involved in all the five conditions of study. Analysis of correlations unveiled the existence of two clusters of related psychiatric conditions, SCZ and BD that were separate from the other three traits, and formed another group. Our results may provide a new insight for genetic basis of the five psychiatric disorders. -
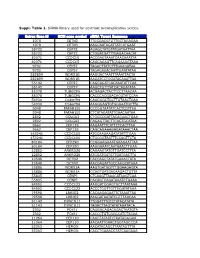
Suppl. Table 1
Suppl. Table 1. SiRNA library used for centriole overduplication screen. Entrez Gene Id NCBI gene symbol siRNA Target Sequence 1070 CETN3 TTGCGACGTGTTGCTAGAGAA 1070 CETN3 AAGCAATAGATTATCATGAAT 55722 CEP72 AGAGCTATGTATGATAATTAA 55722 CEP72 CTGGATGATTTGAGACAACAT 80071 CCDC15 ACCGAGTAAATCAACAAATTA 80071 CCDC15 CAGCAGAGTTCAGAAAGTAAA 9702 CEP57 TAGACTTATCTTTGAAGATAA 9702 CEP57 TAGAGAAACAATTGAATATAA 282809 WDR51B AAGGACTAATTTAAATTACTA 282809 WDR51B AAGATCCTGGATACAAATTAA 55142 CEP27 CAGCAGATCACAAATATTCAA 55142 CEP27 AAGCTGTTTATCACAGATATA 85378 TUBGCP6 ACGAGACTACTTCCTTAACAA 85378 TUBGCP6 CACCCACGGACACGTATCCAA 54930 C14orf94 CAGCGGCTGCTTGTAACTGAA 54930 C14orf94 AAGGGAGTGTGGAAATGCTTA 5048 PAFAH1B1 CCCGGTAATATCACTCGTTAA 5048 PAFAH1B1 CTCATAGATATTGAACAATAA 2802 GOLGA3 CTGGCCGATTACAGAACTGAA 2802 GOLGA3 CAGAGTTACTTCAGTGCATAA 9662 CEP135 AAGAATTTCATTCTCACTTAA 9662 CEP135 CAGCAGAAAGAGATAAACTAA 153241 CCDC100 ATGCAAGAAGATATATTTGAA 153241 CCDC100 CTGCGGTAATTTCCAGTTCTA 80184 CEP290 CCGGAAGAAATGAAGAATTAA 80184 CEP290 AAGGAAATCAATAAACTTGAA 22852 ANKRD26 CAGAAGTATGTTGATCCTTTA 22852 ANKRD26 ATGGATGATGTTGATGACTTA 10540 DCTN2 CACCAGCTATATGAAACTATA 10540 DCTN2 AACGAGATTGCCAAGCATAAA 25886 WDR51A AAGTGATGGTTTGGAAGAGTA 25886 WDR51A CCAGTGATGACAAGACTGTTA 55835 CENPJ CTCAAGTTAAACATAAGTCAA 55835 CENPJ CACAGTCAGATAAATCTGAAA 84902 CCDC123 AAGGATGGAGTGCTTAATAAA 84902 CCDC123 ACCCTGGTTGTTGGATATAAA 79598 LRRIQ2 CACAAGAGAATTCTAAATTAA 79598 LRRIQ2 AAGGATAATATCGTTTAACAA 51143 DYNC1LI1 TTGGATTTGTCTATACATATA 51143 DYNC1LI1 TAGACTTAGTATATAAATACA 2302 FOXJ1 CAGGACAGACAGACTAATGTA -

Identification of Key Genes and Pathways for Alzheimer's Disease
Biophys Rep 2019, 5(2):98–109 https://doi.org/10.1007/s41048-019-0086-2 Biophysics Reports RESEARCH ARTICLE Identification of key genes and pathways for Alzheimer’s disease via combined analysis of genome-wide expression profiling in the hippocampus Mengsi Wu1,2, Kechi Fang1, Weixiao Wang1,2, Wei Lin1,2, Liyuan Guo1,2&, Jing Wang1,2& 1 CAS Key Laboratory of Mental Health, Institute of Psychology, Chinese Academy of Sciences, Beijing 100101, China 2 Department of Psychology, University of Chinese Academy of Sciences, Beijing 10049, China Received: 8 August 2018 / Accepted: 17 January 2019 / Published online: 20 April 2019 Abstract In this study, combined analysis of expression profiling in the hippocampus of 76 patients with Alz- heimer’s disease (AD) and 40 healthy controls was performed. The effects of covariates (including age, gender, postmortem interval, and batch effect) were controlled, and differentially expressed genes (DEGs) were identified using a linear mixed-effects model. To explore the biological processes, func- tional pathway enrichment and protein–protein interaction (PPI) network analyses were performed on the DEGs. The extended genes with PPI to the DEGs were obtained. Finally, the DEGs and the extended genes were ranked using the convergent functional genomics method. Eighty DEGs with q \ 0.1, including 67 downregulated and 13 upregulated genes, were identified. In the pathway enrichment analysis, the 80 DEGs were significantly enriched in one Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway, GABAergic synapses, and 22 Gene Ontology terms. These genes were mainly involved in neuron, synaptic signaling and transmission, and vesicle metabolism. These processes are all linked to the pathological features of AD, demonstrating that the GABAergic system, neurons, and synaptic function might be affected in AD. -

An Exome-Wide Sequencing Study of Lipid Response to High-Fat Meal and Fenofibrate in Caucasians from the GOLDN Cohort
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2018 An exome-wide sequencing study of lipid response to high-fat meal and fenofibrate in Caucasians from the GOLDN cohort Xin Geng Ping An Mary F. Feitosa Michael A. Province et al. Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Supplemental Material can be found at: http://www.jlr.org/content/suppl/2018/02/20/jlr.P080333.DC1 .html patient-oriented and epidemiological research An exome-wide sequencing study of lipid response to high-fat meal and fenofibrate in Caucasians from the GOLDN cohort Xin Geng,* Marguerite R. Irvin,† Bertha Hidalgo,† Stella Aslibekyan,† Vinodh Srinivasasainagendra,§ Ping An,‡ Alexis C. Frazier-Wood,|| Hemant K. Tiwari,§ Tushar Dave,# Kathleen Ryan,# Jose M. Ordovas,$,**,†† Robert J. Straka,§§ Mary F. Feitosa,‡ Paul N. Hopkins,‡‡ Ingrid Borecki,|| || Michael A. Province,‡ Braxton D. Mitchell,# Donna K. Arnett,1,## and Degui Zhi1,*,$$ School of Biomedical Informatics* and School of Public Health,$$ The University of Texas Health Downloaded from Science Center at Houston, Houston, TX 77030; Departments of Epidemiology† and Biostatistics,§ University of Alabama at Birmingham, Birmingham, AL 35233; Division of Statistical Genomics, Department of Genetics,‡ Washington University School of Medicine, St. Louis, MO 63110; US Department of Agriculture/Agricultural Research Service Children’s Nutrition Research Center,|| Baylor College of Medicine, Houston, TX 77030; Department of Medicine, Division -

Whole Exome Sequencing Identifies Causative Mutations in the Majority of Consanguineous Or Familial Cases with Childhood-Onset I
HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Kidney Manuscript Author Int. Author manuscript; Manuscript Author available in PMC 2016 August 01. Published in final edited form as: Kidney Int. 2016 February ; 89(2): 468–475. doi:10.1038/ki.2015.317. Whole exome sequencing identifies causative mutations in the majority of consanguineous or familial cases with childhood- onset increased renal echogenicity Daniela A. Braun#1, Markus Schueler#1, Jan Halbritter1, Heon Yung Gee1, Jonathan D. Porath1, Jennifer A. Lawson1, Rannar Airik1, Shirlee Shril1, Susan J. Allen2, Deborah Stein1, Adila Al Kindy3, Bodo B. Beck4, Nurcan Cengiz5, Khemchand N. Moorani6, Fatih Ozaltin7,8,9, Seema Hashmi10, John A. Sayer11, Detlef Bockenhauer12, Neveen A. Soliman13,14, Edgar A. Otto2, Richard P. Lifton15,16,17, and Friedhelm Hildebrandt1,17 1Division of Nephrology, Department of Medicine, Boston Children's Hospital, Harvard Medical School, Boston, Massachusetts, USA 2Department of Pediatrics, University of Michigan, Michigan, USA 3Department of Genetics, Sultan Qaboos University Hospital, Sultanate of Oman 4Institute for Human Genetics, University of Cologne, Germany 5Baskent University, School of Medicine, Adana Medical Training and Research Center, Department of Pediatric Nephrology, Adana, Turkey 6Department of Pediatric Nephrology, National Institute of Child Health, Karachi 75510, Pakistan 7Faculty of Medicine, Department of Pediatric Nephrology, Hacettepe University, Ankara, Turkey 8Nephrogenetics Laboratory, Faculty of Medicine, -

University of California, San Diego
UC San Diego UC San Diego Electronic Theses and Dissertations Title The post-terminal differentiation fate of RNAs revealed by next-generation sequencing Permalink https://escholarship.org/uc/item/7324r1rj Author Lefkowitz, Gloria Kuo Publication Date 2012 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA, SAN DIEGO The post-terminal differentiation fate of RNAs revealed by next-generation sequencing A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Biomedical Sciences by Gloria Kuo Lefkowitz Committee in Charge: Professor Benjamin D. Yu, Chair Professor Richard Gallo Professor Bruce A. Hamilton Professor Miles F. Wilkinson Professor Eugene Yeo 2012 Copyright Gloria Kuo Lefkowitz, 2012 All rights reserved. The Dissertation of Gloria Kuo Lefkowitz is approved, and it is acceptable in quality and form for publication on microfilm and electronically: __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ Chair University of California, San Diego 2012 iii DEDICATION Ma and Ba, for your early indulgence and support. Matt and James, for choosing more practical callings. Roy, my love, for patiently sharing the ups and downs -

TALPID3 and ANKRD26 Selectively Orchestrate FBF1 Localization and Cilia Gating
ARTICLE https://doi.org/10.1038/s41467-020-16042-w OPEN TALPID3 and ANKRD26 selectively orchestrate FBF1 localization and cilia gating Hao Yan 1,2,3,7, Chuan Chen4,7, Huicheng Chen1,2,3, Hui Hong 1, Yan Huang4,5, Kun Ling4,5, ✉ ✉ Jinghua Hu4,5,6 & Qing Wei 3 Transition fibers (TFs) regulate cilia gating and make the primary cilium a distinct functional entity. However, molecular insights into the biogenesis of a functional cilia gate remain 1234567890():,; elusive. In a forward genetic screen in Caenorhabditis elegans, we uncover that TALP-3, a homolog of the Joubert syndrome protein TALPID3, is a TF-associated component. Genetic analysis reveals that TALP-3 coordinates with ANKR-26, the homolog of ANKRD26, to orchestrate proper cilia gating. Mechanistically, TALP-3 and ANKR-26 form a complex with key gating component DYF-19, the homolog of FBF1. Co-depletion of TALP-3 and ANKR-26 specifically impairs the recruitment of DYF-19 to TFs. Interestingly, in mammalian cells, TALPID3 and ANKRD26 also play a conserved role in coordinating the recruitment of FBF1 to TFs. We thus report a conserved protein module that specifically regulates the functional component of the ciliary gate and suggest a correlation between defective gating and cilio- pathy pathogenesis. 1 CAS Key Laboratory of Insect Developmental and Evolutionary Biology, CAS Center for Excellence in Molecular Plant Sciences, Institute of Plant Physiology and Ecology, Chinese Academy of Sciences, Shanghai 200032, China. 2 University of Chinese Academy of Sciences, Beijing 100039, China. 3 Center for Reproduction and Health Development, Institute of Biomedicine and Biotechnology, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences (CAS), Shenzhen 518055, China.