TALPID3 and ANKRD26 Selectively Orchestrate FBF1 Localization and Cilia Gating
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Old Data and Friends Improve with Age: Advancements with the Updated Tools of Genenetwork
bioRxiv preprint doi: https://doi.org/10.1101/2021.05.24.445383; this version posted May 25, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Old data and friends improve with age: Advancements with the updated tools of GeneNetwork Alisha Chunduri1, David G. Ashbrook2 1Department of Biotechnology, Chaitanya Bharathi Institute of Technology, Hyderabad 500075, India 2Department of Genetics, Genomics and Informatics, University of Tennessee Health Science Center, Memphis, TN 38163, USA Abstract Understanding gene-by-environment interactions is important across biology, particularly behaviour. Families of isogenic strains are excellently placed, as the same genome can be tested in multiple environments. The BXD’s recent expansion to 140 strains makes them the largest family of murine isogenic genomes, and therefore give great power to detect QTL. Indefinite reproducible genometypes can be leveraged; old data can be reanalysed with emerging tools to produce novel biological insights. To highlight the importance of reanalyses, we obtained drug- and behavioural-phenotypes from Philip et al. 2010, and reanalysed their data with new genotypes from sequencing, and new models (GEMMA and R/qtl2). We discover QTL on chromosomes 3, 5, 9, 11, and 14, not found in the original study. We narrowed down the candidate genes based on their ability to alter gene expression and/or protein function, using cis-eQTL analysis, and variants predicted to be deleterious. Co-expression analysis (‘gene friends’) and human PheWAS were used to further narrow candidates. -

The Hydrolethalus Syndrome Protein HYLS-1 Regulates Formation of the Ciliary Gate
ARTICLE Received 8 Sep 2015 | Accepted 30 Jun 2016 | Published 18 Aug 2016 DOI: 10.1038/ncomms12437 OPEN The hydrolethalus syndrome protein HYLS-1 regulates formation of the ciliary gate Qing Wei1,2,*, Yingyi Zhang1,*, Clementine Schouteden3, Yuxia Zhang1, Qing Zhang1, Jinhong Dong1, Veronika Wonesch3, Kun Ling1, Alexander Dammermann3 & Jinghua Hu1,4,5 Transition fibres (TFs), together with the transition zone (TZ), are basal ciliary structures thought to be crucial for cilium biogenesis and function by acting as a ciliary gate to regulate selective protein entry and exit. Here we demonstrate that the centriolar and basal body protein HYLS-1, the C. elegans orthologue of hydrolethalus syndrome protein 1, is required for TF formation, TZ organization and ciliary gating. Loss of HYLS-1 compromises the docking and entry of intraflagellar transport (IFT) particles, ciliary gating for both membrane and soluble proteins, and axoneme assembly. Additional depletion of the TF component DYF-19 in hyls-1 mutants further exacerbates TZ anomalies and completely abrogates ciliogenesis. Our data support an important role for HYLS-1 and TFs in establishment of the ciliary gate and underline the importance of selective protein entry for cilia assembly. 1 Department of Biochemistry and Molecular Biology, Mayo Clinic, Rochester, Minnesota 55905, USA. 2 Key Laboratory of Insect Developmental and Evolutionary Biology, Institute of Plant Physiology and Ecology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai 200032, China. 3 Max F. Perutz Laboratories, Vienna Biocenter (VBC), University of Vienna, A-1030 Vienna, Austria. 4 Division of Nephrology and Hypertension, Mayo Clinic, Rochester, Minnesota 55905, USA. 5 Mayo Translational PKD Center, Mayo Clinic, Rochester, Minnesota 55905, USA. -

Unraveling the Genetics of Joubert and Meckel-Gruber Syndromes
Journal of Pediatric Genetics 3 (2014) 65–78 65 DOI 10.3233/PGE-14090 IOS Press Unraveling the genetics of Joubert and Meckel-Gruber syndromes Katarzyna Szymanska, Verity L. Hartill and Colin A. Johnson∗ Department of Ophthalmology and Neuroscience, University of Leeds, Leeds, UK Received 27 May 2014 Revised 11 July 2014 Accepted 14 July 2014 Abstract. Joubert syndrome (JBTS) and Meckel-Gruber syndrome (MKS) are recessive neurodevelopmental conditions caused by mutations in proteins that are structural or functional components of the primary cilium. In this review, we provide an overview of their clinical diagnosis, management and molecular genetics. Both have variable phenotypes, extreme genetic heterogeneity, and display allelism both with each other and other ciliopathies. Recent advances in genetic technology have significantly improved diagnosis and clinical management of ciliopathy patients, with the delineation of some general genotype-phenotype correlations. We highlight those that are most relevant for clinical practice, including the correlation between TMEM67 mutations and the JBTS variant phenotype of COACH syndrome. The subcellular localization of the known MKS and JBTS proteins is now well-described, and we discuss some of the contemporary ideas about ciliopathy disease pathogenesis. Most JBTS and MKS proteins localize to a discrete ciliary compartment called the transition zone, and act as structural components of the so-called “ciliary gate” to regulate the ciliary trafficking of cargo proteins or lipids. Cargo proteins include enzymes and transmembrane proteins that mediate intracellular signaling. The disruption of transition zone function may contribute to the ciliopathy phenotype by altering the composition of the ciliary membrane or axoneme, with impacts on essential developmental signaling including the Wnt and Shh pathways as well as the regulation of secondary messengers such as inositol-1,4,5-trisphosphate (InsP3) and cyclic adenosine monophosphate (cAMP). -

Synergistic Genetic Interactions Between Pkhd1 and Pkd1 Result in an ARPKD-Like Phenotype in Murine Models
BASIC RESEARCH www.jasn.org Synergistic Genetic Interactions between Pkhd1 and Pkd1 Result in an ARPKD-Like Phenotype in Murine Models Rory J. Olson,1 Katharina Hopp ,2 Harrison Wells,3 Jessica M. Smith,3 Jessica Furtado,1,4 Megan M. Constans,3 Diana L. Escobar,3 Aron M. Geurts,5 Vicente E. Torres,3 and Peter C. Harris 1,3 Due to the number of contributing authors, the affiliations are listed at the end of this article. ABSTRACT Background Autosomal recessive polycystic kidney disease (ARPKD) and autosomal dominant polycystic kidney disease (ADPKD) are genetically distinct, with ADPKD usually caused by the genes PKD1 or PKD2 (encoding polycystin-1 and polycystin-2, respectively) and ARPKD caused by PKHD1 (encoding fibrocys- tin/polyductin [FPC]). Primary cilia have been considered central to PKD pathogenesis due to protein localization and common cystic phenotypes in syndromic ciliopathies, but their relevance is questioned in the simple PKDs. ARPKD’s mild phenotype in murine models versus in humans has hampered investi- gating its pathogenesis. Methods To study the interaction between Pkhd1 and Pkd1, including dosage effects on the phenotype, we generated digenic mouse and rat models and characterized and compared digenic, monogenic, and wild-type phenotypes. Results The genetic interaction was synergistic in both species, with digenic animals exhibiting pheno- types of rapidly progressive PKD and early lethality resembling classic ARPKD. Genetic interaction be- tween Pkhd1 and Pkd1 depended on dosage in the digenic murine models, with no significant enhancement of the monogenic phenotype until a threshold of reduced expression at the second locus was breached. -

The Basal Bodies of Chlamydomonas Reinhardtii Susan K
Dutcher and O’Toole Cilia (2016) 5:18 DOI 10.1186/s13630-016-0039-z Cilia REVIEW Open Access The basal bodies of Chlamydomonas reinhardtii Susan K. Dutcher1* and Eileen T. O’Toole2 Abstract The unicellular green alga, Chlamydomonas reinhardtii, is a biflagellated cell that can swim or glide. C. reinhardtii cells are amenable to genetic, biochemical, proteomic, and microscopic analysis of its basal bodies. The basal bodies contain triplet microtubules and a well-ordered transition zone. Both the mother and daughter basal bodies assemble flagella. Many of the proteins found in other basal body-containing organisms are present in the Chlamydomonas genome, and mutants in these genes affect the assembly of basal bodies. Electron microscopic analysis shows that basal body duplication is site-specific and this may be important for the proper duplication and spatial organization of these organelles. Chlamydomonas is an excellent model for the study of basal bodies as well as the transition zone. Keywords: Site-specific basal body duplication, Cartwheel, Transition zone, Centrin fibers Phylogeny and conservation of proteins Centrin, SPD2/CEP192, Asterless/CEP152; CEP70, The green lineage or Viridiplantae consists of the green delta-tubulin, and epsilon-tubulin. Chlamydomonas has algae, which include Chlamydomonas, the angiosperms homologs of all of these based on sequence conservation (the land plants), and the gymnosperms (conifers, cycads, except PLK4, CEP152, and CEP192. Several lines of evi- ginkgos). They are grouped together because they have dence suggests that CEP152, CEP192, and PLK4 interact chlorophyll a and b and lack phycobiliproteins. The green [20, 52] and their concomitant absence in several organ- algae together with the cycads and ginkgos have basal isms suggests that other mechanisms exist that allow for bodies and cilia, while the angiosperms and conifers have control of duplication [4]. -

Par6c Is at the Mother Centriole and Controls Centrosomal Protein
860 Research Article Par6c is at the mother centriole and controls centrosomal protein composition through a Par6a-dependent pathway Vale´rian Dormoy, Kati Tormanen and Christine Su¨ tterlin* Department of Developmental and Cell Biology, University of California, Irvine, Irvine, CA 92697-2300, USA *Author for correspondence ([email protected]) Accepted 3 December 2012 Journal of Cell Science 126, 860–870 ß 2013. Published by The Company of Biologists Ltd doi: 10.1242/jcs.121186 Summary The centrosome contains two centrioles that differ in age, protein composition and function. This non-membrane bound organelle is known to regulate microtubule organization in dividing cells and ciliogenesis in quiescent cells. These specific roles depend on protein appendages at the older, or mother, centriole. In this study, we identified the polarity protein partitioning defective 6 homolog gamma (Par6c) as a novel component of the mother centriole. This specific localization required the Par6c C-terminus, but was independent of intact microtubules, the dynein/dynactin complex and the components of the PAR polarity complex. Par6c depletion resulted in altered centrosomal protein composition, with the loss of a large number of proteins, including Par6a and p150Glued, from the centrosome. As a consequence, there were defects in ciliogenesis, microtubule organization and centrosome reorientation during migration. Par6c interacted with Par3 and aPKC, but these proteins were not required for the regulation of centrosomal protein composition. Par6c also associated with Par6a, which controls protein recruitment to the centrosome through p150Glued. Our study is the first to identify Par6c as a component of the mother centriole and to report a role of a mother centriole protein in the regulation of centrosomal protein composition. -

Joubert Syndrome Genereview
Title: Joubert Syndrome GeneReview — Molecular Genetics: Less Common Genetic Causes Authors: Parisi M, Glass I Updated: June 2017 Note: The following information is provided by the authors listed above and has not been reviewed by GeneReviews staff. Joubert Syndrome: Less Common Genetic Causes ARL13B B9D1 B9D2 CEP41 IFT172 KIF7 OFD1 (CXORF5) PDE6D POC1B TCTN1 TCTN3 TMEM138 TMEM231 TMEM237 (ALS2CR4) TTC21B ARL13B Gene structure. ARL13B is a ten-exon gene that encodes a 428-amino acid protein. Pathogenic variants. Two families with a phenotype typical of classic Joubert syndrome had missense and/or nonsense variants in this gene; one of these individuals also had evidence of a retinopathy [Cantagrel et al 2008]. Normal gene product. ARL13B encodes ADP-ribosylation factor-like protein 13B, a member of the ADP-ribosylation factor-like family. Multiple transcript variants result from alternate splicing; two protein isoforms are known. The AR13B protein is a small GTPase in the Ras superfamily that contains both N-terminal and C-terminal guanine nucleotide-binding motifs. It is localized to the cilia and plays a role in cilia formation and maintenance as well as sonic hedgehog signaling. Abnormal gene product. In C elegans, pathogenic variants in the homolog arl13 exhibit defective cilium morphology, localization, and anterograde intraflagellar transport [Cevik et al 2010]. Mice with defects in the murine ortholog have neural tube defects and polydactyly, as well as an embryonic-lethal phenotype [Cantagrel et al 2008, Doherty 2009]. B9D1. See Tables A and B. B9D2. See Tables A and B. CEP41 Gene structure. The gene consists of 11 exons and spans approximately 50 kb. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Pallister–Hall Syndrome
1-10-2020 Pallister–Hall Syndrome J Pediatr Neurosci. 2017 Jul-Sep; 12(3): 276–279. PMCID: PMC5696670 doi: 10.4103/jpn.JPN_101_17: 10.4103/jpn.JPN_101_17 PMID: 29204208 Pallister–Hall Syndrome Sadanandvalli Retnaswami Chandra, Mane Maheshkumar Daryappa,1 M. A. Mukheem Mudabbir, 1 M. Pooja, and A. Arivazhagan Neurocentre, National Institute of Mental Health and Neurosciences, Bengaluru, Karnataka, India 1Department of Neurology, National Institute of Mental Health and Neurosciences, Bengaluru, Karnataka, India Address for correspondence: Dr. Sadanandavalli Retnaswami Chandra, Department of Neurology, National Institute of Mental Health and Neurosciences, Bangalore, Karnataka, India. E-mail: [email protected] Copyright : © 2017 Journal of Pediatric Neurosciences This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial- ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms. Abstract Polydactyly is a relatively common abnormality in infants. However, it can be a marker of a wide variety of neurological and systemic abnormality. Hence, it is important for pediatrician and physician to have insight into the various association of this apparently innocuous anomaly. In this write-up, we report an extremely rare syndrome associated with polydactyly that is Pallister–Hall syndrome. A 10-month-old male child born by lower segment cesarean section presented with global delay associated with microcephaly, frontal bossing, hypertelorism, flat nose, short philtrum, incomplete cleft in the upper lip and hard palate, polydactyly, and syndactyly. The child presented with repeated vomiting and crying episodes. -

Mitochondrial Medicine in the Omics Era
Mitochondrial Medicine in the Omics Era Joyeeta Rahman1 and Shamima Rahman1,2* 1 Mitochondrial Research Group, UCL Great Ormond Street Institute of Child Health and 2 Metabolic Unit, Great Ormond Street Hospital NHS Foundation Trust, London, UK *Correspondence to: Professor Shamima Rahman Mitochondrial Research Group Genetics and Genomic Medicine UCL Great Ormond Street Institute of Child Health London WC1N 1EH, UK. Telephone: +44 (0)2079052608 [email protected] Keywords: Mitochondrial disease, OXPHOS, signalling, omics, genomics, transcriptomics, proteomics, metabolomics, mitochondrial stress response, treatment 1 Abstract Mitochondria are dynamic bioenergetic organelles whose maintenance requires ~1500 proteins from two genomes. Mutations in either the mitochondrial or nuclear genome can disrupt a plethora of cellular metabolic and homeostatic functions. Mitochondrial diseases represent one the most common and severe groups of inherited genetic disorders, characterised by clinical, biochemical, and genetic heterogeneity, diagnostic odysseys, and lack of curative therapies. This review aims to discuss recent advances in mitochondrial biology and medicine arising from widespread utilisation of high-throughput omics technologies, and also includes a broad discussion of emerging therapies for mitochondrial disease. New insights into both bioenergetic and biosynthetic mitochondrial functionalities have expedited the genetic diagnosis of primary mitochondrial disorders, and identified novel mitochondrial pathomechanisms and new targets -

The Transformation of the Centrosome Into the Basal Body: Similarities and Dissimilarities Between Somatic and Male Germ Cells and Their Relevance for Male Fertility
cells Review The Transformation of the Centrosome into the Basal Body: Similarities and Dissimilarities between Somatic and Male Germ Cells and Their Relevance for Male Fertility Constanza Tapia Contreras and Sigrid Hoyer-Fender * Göttingen Center of Molecular Biosciences, Johann-Friedrich-Blumenbach Institute for Zoology and Anthropology-Developmental Biology, Faculty of Biology and Psychology, Georg-August University of Göttingen, 37077 Göttingen, Germany; [email protected] * Correspondence: [email protected] Abstract: The sperm flagellum is essential for the transport of the genetic material toward the oocyte and thus the transmission of the genetic information to the next generation. During the haploid phase of spermatogenesis, i.e., spermiogenesis, a morphological and molecular restructuring of the male germ cell, the round spermatid, takes place that includes the silencing and compaction of the nucleus, the formation of the acrosomal vesicle from the Golgi apparatus, the formation of the sperm tail, and, finally, the shedding of excessive cytoplasm. Sperm tail formation starts in the round spermatid stage when the pair of centrioles moves toward the posterior pole of the nucleus. The sperm tail, eventually, becomes located opposed to the acrosomal vesicle, which develops at the anterior pole of the nucleus. The centriole pair tightly attaches to the nucleus, forming a nuclear membrane indentation. An Citation: Tapia Contreras, C.; articular structure is formed around the centriole pair known as the connecting piece, situated in the Hoyer-Fender, S. The Transformation neck region and linking the sperm head to the tail, also named the head-to-tail coupling apparatus or, of the Centrosome into the Basal in short, HTCA. -
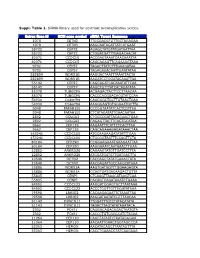
Suppl. Table 1
Suppl. Table 1. SiRNA library used for centriole overduplication screen. Entrez Gene Id NCBI gene symbol siRNA Target Sequence 1070 CETN3 TTGCGACGTGTTGCTAGAGAA 1070 CETN3 AAGCAATAGATTATCATGAAT 55722 CEP72 AGAGCTATGTATGATAATTAA 55722 CEP72 CTGGATGATTTGAGACAACAT 80071 CCDC15 ACCGAGTAAATCAACAAATTA 80071 CCDC15 CAGCAGAGTTCAGAAAGTAAA 9702 CEP57 TAGACTTATCTTTGAAGATAA 9702 CEP57 TAGAGAAACAATTGAATATAA 282809 WDR51B AAGGACTAATTTAAATTACTA 282809 WDR51B AAGATCCTGGATACAAATTAA 55142 CEP27 CAGCAGATCACAAATATTCAA 55142 CEP27 AAGCTGTTTATCACAGATATA 85378 TUBGCP6 ACGAGACTACTTCCTTAACAA 85378 TUBGCP6 CACCCACGGACACGTATCCAA 54930 C14orf94 CAGCGGCTGCTTGTAACTGAA 54930 C14orf94 AAGGGAGTGTGGAAATGCTTA 5048 PAFAH1B1 CCCGGTAATATCACTCGTTAA 5048 PAFAH1B1 CTCATAGATATTGAACAATAA 2802 GOLGA3 CTGGCCGATTACAGAACTGAA 2802 GOLGA3 CAGAGTTACTTCAGTGCATAA 9662 CEP135 AAGAATTTCATTCTCACTTAA 9662 CEP135 CAGCAGAAAGAGATAAACTAA 153241 CCDC100 ATGCAAGAAGATATATTTGAA 153241 CCDC100 CTGCGGTAATTTCCAGTTCTA 80184 CEP290 CCGGAAGAAATGAAGAATTAA 80184 CEP290 AAGGAAATCAATAAACTTGAA 22852 ANKRD26 CAGAAGTATGTTGATCCTTTA 22852 ANKRD26 ATGGATGATGTTGATGACTTA 10540 DCTN2 CACCAGCTATATGAAACTATA 10540 DCTN2 AACGAGATTGCCAAGCATAAA 25886 WDR51A AAGTGATGGTTTGGAAGAGTA 25886 WDR51A CCAGTGATGACAAGACTGTTA 55835 CENPJ CTCAAGTTAAACATAAGTCAA 55835 CENPJ CACAGTCAGATAAATCTGAAA 84902 CCDC123 AAGGATGGAGTGCTTAATAAA 84902 CCDC123 ACCCTGGTTGTTGGATATAAA 79598 LRRIQ2 CACAAGAGAATTCTAAATTAA 79598 LRRIQ2 AAGGATAATATCGTTTAACAA 51143 DYNC1LI1 TTGGATTTGTCTATACATATA 51143 DYNC1LI1 TAGACTTAGTATATAAATACA 2302 FOXJ1 CAGGACAGACAGACTAATGTA