Unraveling the Genetics of Joubert and Meckel-Gruber Syndromes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Educational Paper Ciliopathies
Eur J Pediatr (2012) 171:1285–1300 DOI 10.1007/s00431-011-1553-z REVIEW Educational paper Ciliopathies Carsten Bergmann Received: 11 June 2011 /Accepted: 3 August 2011 /Published online: 7 September 2011 # The Author(s) 2011. This article is published with open access at Springerlink.com Abstract Cilia are antenna-like organelles found on the (NPHP) . Ivemark syndrome . Meckel syndrome (MKS) . surface of most cells. They transduce molecular signals Joubert syndrome (JBTS) . Bardet–Biedl syndrome (BBS) . and facilitate interactions between cells and their Alstrom syndrome . Short-rib polydactyly syndromes . environment. Ciliary dysfunction has been shown to Jeune syndrome (ATD) . Ellis-van Crefeld syndrome (EVC) . underlie a broad range of overlapping, clinically and Sensenbrenner syndrome . Primary ciliary dyskinesia genetically heterogeneous phenotypes, collectively (Kartagener syndrome) . von Hippel-Lindau (VHL) . termed ciliopathies. Literally, all organs can be affected. Tuberous sclerosis (TSC) . Oligogenic inheritance . Modifier. Frequent cilia-related manifestations are (poly)cystic Mutational load kidney disease, retinal degeneration, situs inversus, cardiac defects, polydactyly, other skeletal abnormalities, and defects of the central and peripheral nervous Introduction system, occurring either isolated or as part of syn- dromes. Characterization of ciliopathies and the decisive Defective cellular organelles such as mitochondria, perox- role of primary cilia in signal transduction and cell isomes, and lysosomes are well-known -
Shimada 2017.Pdf
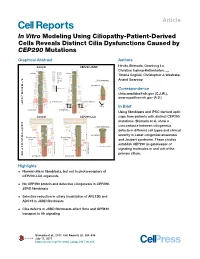
Article In Vitro Modeling Using Ciliopathy-Patient-Derived Cells Reveals Distinct Cilia Dysfunctions Caused by CEP290 Mutations Graphical Abstract Authors Control CEP290-JSRD Hiroko Shimada, Quanlong Lu, Christine Insinna-Kettenhofen, ..., Cilium GPR161 Cilium ADCY3 Tiziana Cogliati, Christopher J. Westlake, Smo ARL13B F CEP290 Cell membrane Anand Swaroop I Gli2 B Cytoplasm R Other proteins O Correspondence Axoneme B Axoneme [email protected] (C.J.W.), L A [email protected] (A.S.) S Cell membrane Cell membrane T Vesicles In Brief Cytoplasm Cytoplasm Using fibroblasts and iPSC-derived optic Control CEP290-LCA cups from patients with distinct CEP290 mutations, Shimada et al. show a P H Cell membrane concordance between ciliogenesis O T ? defects in different cell types and clinical Axoneme ? O ? Axoneme ? Cytoplasm severity in Leber congenital amaurosis R ? Connecting Connecting ? E Cilium Cilium ? and Joubert syndrome. These studies Cell membrane C Cell membrane E establish CEP290 as gatekeeper of P Docked mother centriole T ?? ? ? signaling molecules in and out of the O primary cilium. R Cytoplasm Cytoplasm Highlights d Normal cilia in fibroblasts, but not in photoreceptors of CEP290-LCA organoids d No CEP290 protein and defective ciliogenesis in CEP290- JSRD fibroblasts d Selective reduction in ciliary localization of ARL13B and ADCY3 in JSRD fibroblasts d Cilia defects in JSRD fibroblasts affect Smo and GPR161 transport in Hh signaling Shimada et al., 2017, Cell Reports 20, 384–396 July 11, 2017 http://dx.doi.org/10.1016/j.celrep.2017.06.045 Cell Reports Article In Vitro Modeling Using Ciliopathy-Patient-Derived Cells Reveals Distinct Cilia Dysfunctions Caused by CEP290 Mutations Hiroko Shimada,1 Quanlong Lu,2 Christine Insinna-Kettenhofen,2 Kunio Nagashima,3 Milton A. -

The Hydrolethalus Syndrome Protein HYLS-1 Regulates Formation of the Ciliary Gate
ARTICLE Received 8 Sep 2015 | Accepted 30 Jun 2016 | Published 18 Aug 2016 DOI: 10.1038/ncomms12437 OPEN The hydrolethalus syndrome protein HYLS-1 regulates formation of the ciliary gate Qing Wei1,2,*, Yingyi Zhang1,*, Clementine Schouteden3, Yuxia Zhang1, Qing Zhang1, Jinhong Dong1, Veronika Wonesch3, Kun Ling1, Alexander Dammermann3 & Jinghua Hu1,4,5 Transition fibres (TFs), together with the transition zone (TZ), are basal ciliary structures thought to be crucial for cilium biogenesis and function by acting as a ciliary gate to regulate selective protein entry and exit. Here we demonstrate that the centriolar and basal body protein HYLS-1, the C. elegans orthologue of hydrolethalus syndrome protein 1, is required for TF formation, TZ organization and ciliary gating. Loss of HYLS-1 compromises the docking and entry of intraflagellar transport (IFT) particles, ciliary gating for both membrane and soluble proteins, and axoneme assembly. Additional depletion of the TF component DYF-19 in hyls-1 mutants further exacerbates TZ anomalies and completely abrogates ciliogenesis. Our data support an important role for HYLS-1 and TFs in establishment of the ciliary gate and underline the importance of selective protein entry for cilia assembly. 1 Department of Biochemistry and Molecular Biology, Mayo Clinic, Rochester, Minnesota 55905, USA. 2 Key Laboratory of Insect Developmental and Evolutionary Biology, Institute of Plant Physiology and Ecology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai 200032, China. 3 Max F. Perutz Laboratories, Vienna Biocenter (VBC), University of Vienna, A-1030 Vienna, Austria. 4 Division of Nephrology and Hypertension, Mayo Clinic, Rochester, Minnesota 55905, USA. 5 Mayo Translational PKD Center, Mayo Clinic, Rochester, Minnesota 55905, USA. -

Prenatal Exome Sequencing in Anomalous Fetuses: New Opportunities and Challenges
© American College of Medical Genetics and Genomics ORIGINAL RESEARCH ARTICLE Prenatal exome sequencing in anomalous fetuses: new opportunities and challenges Neeta L. Vora, MD1, Bradford Powell, MD, PhD2, Alicia Brandt, MS2, Natasha Strande, PhD3,4, Emily Hardisty, MS, CGC1, Kelly Gilmore, MS, CGC1, Ann Katherine M. Foreman, MS, CGC2,5, Kirk Wilhelmsen, MD, PhD6, Chris Bizon, PhD6, Jason Reilly, BA6, Phil Owen, BS6, Cynthia M. Powell, MD, MS2,7, Debra Skinner, PhD, MA8, Christine Rini, PhD9, Anne D. Lyerly, MD, MA10, Kim A. Boggess, MD1, Karen Weck, MD3,4, Jonathan S. Berg, MD, PhD2 and James P. Evans, MD, PhD2,10 Purpose: We investigated the diagnostic and clinical performance the following genes: COL1A1, MUSK, KCTD1, RTTN, TMEM67, of exome sequencing in fetuses with sonographic abnormalities PIEZO1 and DYNC2H1. One additional case revealed a de novo with normal karyotype and microarray and, in some cases, normal nonsense mutation in a novel candidate gene (MAP4K4). gene-specific sequencing. The perceived likelihood that exome sequencing would explain Methods: Exome sequencing was performed on DNA from 15 the results (5.2 on a 10-point scale) was higher than the anomalous fetuses and from the peripheral blood of their approximately 30% diagnostic yield discussed in pretest counseling. parents. Parents provided consent to be informed of diagnostic Conclusion: Exome sequencing had diagnostic utility in a highly results in the fetus, medically actionable findings in the parents, select population of fetuses where a genetic diagnosis was highly and their identification as carrier couples for significant auto- suspected. Challenges related to genetics literacy and variant somal recessive conditions. -

Ciliopathies Gene Panel
Ciliopathies Gene Panel Contact details Introduction Regional Genetics Service The ciliopathies are a heterogeneous group of conditions with considerable phenotypic overlap. Levels 4-6, Barclay House These inherited diseases are caused by defects in cilia; hair-like projections present on most 37 Queen Square cells, with roles in key human developmental processes via their motility and signalling functions. Ciliopathies are often lethal and multiple organ systems are affected. Ciliopathies are London, WC1N 3BH united in being genetically heterogeneous conditions and the different subtypes can share T +44 (0) 20 7762 6888 many clinical features, predominantly cystic kidney disease, but also retinal, respiratory, F +44 (0) 20 7813 8578 skeletal, hepatic and neurological defects in addition to metabolic defects, laterality defects and polydactyly. Their clinical variability can make ciliopathies hard to recognise, reflecting the ubiquity of cilia. Gene panels currently offer the best solution to tackling analysis of genetically Samples required heterogeneous conditions such as the ciliopathies. Ciliopathies affect approximately 1:2,000 5ml venous blood in plastic EDTA births. bottles (>1ml from neonates) Ciliopathies are generally inherited in an autosomal recessive manner, with some autosomal Prenatal testing must be arranged dominant and X-linked exceptions. in advance, through a Clinical Genetics department if possible. Referrals Amniotic fluid or CV samples Patients presenting with a ciliopathy; due to the phenotypic variability this could be a diverse set should be sent to Cytogenetics for of features. For guidance contact the laboratory or Dr Hannah Mitchison dissecting and culturing, with ([email protected]) / Prof Phil Beales ([email protected]) instructions to forward the sample to the Regional Molecular Genetics Referrals will be accepted from clinical geneticists and consultants in nephrology, metabolic, laboratory for analysis respiratory and retinal diseases. -

Joubert Syndrome Genereview
Title: Joubert Syndrome GeneReview — Molecular Genetics: Less Common Genetic Causes Authors: Parisi M, Glass I Updated: June 2017 Note: The following information is provided by the authors listed above and has not been reviewed by GeneReviews staff. Joubert Syndrome: Less Common Genetic Causes ARL13B B9D1 B9D2 CEP41 IFT172 KIF7 OFD1 (CXORF5) PDE6D POC1B TCTN1 TCTN3 TMEM138 TMEM231 TMEM237 (ALS2CR4) TTC21B ARL13B Gene structure. ARL13B is a ten-exon gene that encodes a 428-amino acid protein. Pathogenic variants. Two families with a phenotype typical of classic Joubert syndrome had missense and/or nonsense variants in this gene; one of these individuals also had evidence of a retinopathy [Cantagrel et al 2008]. Normal gene product. ARL13B encodes ADP-ribosylation factor-like protein 13B, a member of the ADP-ribosylation factor-like family. Multiple transcript variants result from alternate splicing; two protein isoforms are known. The AR13B protein is a small GTPase in the Ras superfamily that contains both N-terminal and C-terminal guanine nucleotide-binding motifs. It is localized to the cilia and plays a role in cilia formation and maintenance as well as sonic hedgehog signaling. Abnormal gene product. In C elegans, pathogenic variants in the homolog arl13 exhibit defective cilium morphology, localization, and anterograde intraflagellar transport [Cevik et al 2010]. Mice with defects in the murine ortholog have neural tube defects and polydactyly, as well as an embryonic-lethal phenotype [Cantagrel et al 2008, Doherty 2009]. B9D1. See Tables A and B. B9D2. See Tables A and B. CEP41 Gene structure. The gene consists of 11 exons and spans approximately 50 kb. -

Renal Cystic Disorders Infosheet 6-14-19
Next Generation Sequencing Panel for Renal Cystic Disorders Clinical Features: Renal cystic diseases are a genetically heterogeneous group of conditions characterized By isolated renal disease or renal cysts in conjunction with extrarenal features (1). Age of onset of renal cystic disease ranges from neonatal to adult onset. Common features of renal cystic diseases include renal insufficiency and progression to end stage renal disease (ESRD). Identification of the genetic etiology of renal cystic disease can aid in appropriate clinical management of the affected patient. Our Renal Cystic Disorders Panel includes sequence and deletion/duplicaton analysis of all 79 genes listed below. Renal Cystic Disorders Sequencing Panel AHI1 BMPER HNF1B NEK8 TCTN3 WDPCP ANKS6 C5orf42 IFT27 NOTCH2 TFAP2A WDR19 ARL13B CC2D2A IFT140 NPHP1 TMEM107 XPNPEP3 ARL6 CDC73 IFT172 NPHP3 TMEM138 ZNF423 B9D1 CEP104 INPP5E NPHP4 TMEM216 B9D2 CEP120 INVS OFD1 TMEM231 BBIP1 CEP164 IQCB1 PDE6D TMEM237 BBS1 CEP290 JAG1 PKD2 TMEM67 BBS10 CEP41 KIAA0556 PKHD1 TRIM32 BBS12 CEP83 KIAA0586 REN TSC1 BBS2 CRB2 KIF14 RPGRIP1L TSC2 BBS4 CSPP1 KIF7 SALL1 TTC21B BBS5 DCDC2 LZTFL1 SDCCAG8 TTC8 BBS7 GLIS2 MKKS TCTN1 UMOD BBS9 GLIS3 MKS1 TCTN2 VHL Disorder Genes Inheritance Clinical features/molecular genetics Bardet Biedl ARL6 AR Bardet-Biedl syndrome (BBS) is an autosomal syndrome BBS1 recessive multi-systemic ciliopathy characterized By BBS10 retinal dystrophy, oBesity, postaxial polydactyly, BBS12 leaning difficulties, renal involvement and BBS2 genitourinary abnormalities (2). Visual prognosis is BBS4 poor, and the mean age of legal Blindness is 15.5 BBS5 years. Birth weight is typically normal But significant BBS7 weight gain Begins within the first year. Renal BBS9 disease is a major cause of morBidity and mortality. -

Pallister–Hall Syndrome
1-10-2020 Pallister–Hall Syndrome J Pediatr Neurosci. 2017 Jul-Sep; 12(3): 276–279. PMCID: PMC5696670 doi: 10.4103/jpn.JPN_101_17: 10.4103/jpn.JPN_101_17 PMID: 29204208 Pallister–Hall Syndrome Sadanandvalli Retnaswami Chandra, Mane Maheshkumar Daryappa,1 M. A. Mukheem Mudabbir, 1 M. Pooja, and A. Arivazhagan Neurocentre, National Institute of Mental Health and Neurosciences, Bengaluru, Karnataka, India 1Department of Neurology, National Institute of Mental Health and Neurosciences, Bengaluru, Karnataka, India Address for correspondence: Dr. Sadanandavalli Retnaswami Chandra, Department of Neurology, National Institute of Mental Health and Neurosciences, Bangalore, Karnataka, India. E-mail: [email protected] Copyright : © 2017 Journal of Pediatric Neurosciences This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial- ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms. Abstract Polydactyly is a relatively common abnormality in infants. However, it can be a marker of a wide variety of neurological and systemic abnormality. Hence, it is important for pediatrician and physician to have insight into the various association of this apparently innocuous anomaly. In this write-up, we report an extremely rare syndrome associated with polydactyly that is Pallister–Hall syndrome. A 10-month-old male child born by lower segment cesarean section presented with global delay associated with microcephaly, frontal bossing, hypertelorism, flat nose, short philtrum, incomplete cleft in the upper lip and hard palate, polydactyly, and syndactyly. The child presented with repeated vomiting and crying episodes. -

Evolving Concepts in Human Renal Dysplasia
DISEASE OF THE MONTH J Am Soc Nephrol 15: 998–1007, 2004 EBERHARD RITZ, FEATURE EDITOR Evolving Concepts in Human Renal Dysplasia ADRIAN S. WOOLF, KAREN L. PRICE, PETER J. SCAMBLER, and PAUL J.D. WINYARD Nephro-Urology and Molecular Medicine Units, Institute of Child Health, University College London, London, United Kingdom Abstract. Human renal dysplasia is a collection of disorders in correlating with perturbed cell turnover and maturation. Mu- which kidneys begin to form but then fail to differentiate into tations of nephrogenesis genes have been defined in multiorgan normal nephrons and collecting ducts. Dysplasia is the princi- dysmorphic disorders in which renal dysplasia can feature, pal cause of childhood end-stage renal failure. Two main including Fraser, renal cysts and diabetes, and Kallmann syn- theories have been considered in its pathogenesis: A primary dromes. Here, it is possible to begin to understand the normal failure of ureteric bud activity and a disruption produced by nephrogenic function of the wild-type proteins and understand fetal urinary flow impairment. Recent studies have docu- how mutations might cause aberrant organogenesis. mented deregulation of gene expression in human dysplasia, Congenital anomalies of the kidney and urinary tract and the main renal pathology is renal dysplasia (RD). In her (CAKUT) account for one third of all anomalies detected by landmark book Normal and Abnormal Development of the routine fetal ultrasonography (1). A recent UK audit of child- Kidney published in 1972 (7), Edith Potter emphasized that one hood end-stage renal failure reported that CAKUT was the must understand normal development to generate realistic hy- cause in ~40% of 882 individuals (2). -

Histone Deacetylase 6 Represents a Novel Drug Target in the Oncogenic Hedgehog Signaling Pathway
Aus dem Institut für Molekularbiologie und Tumorforschung Geschäftsführender Direktor: Prof. Dr. Rolf Müller des Fachbereichs Medizin der Philipps-Universität Marburg Histone deacetylase 6 represents a novel drug target in the oncogenic Hedgehog signaling pathway Inaugural- Dissertation zur Erlangung des Doktorgrades der Naturwissenschaften dem Fachbereich Medizin der Philipps-Universität Marburg vorgelegt von Dhanyamraju Pavan Kumar aus Secunderabad, Indien Marburg-2017 Angenommen vom Fachbereich Medizin der Philipps Universität Marburg am:3/11/2017 Gedruckt mit der Genehmigung des Fachbereichs Dekan: Prof. Dr. Helmut Schäfer Referent: PD. Dr. Matthias Lauth Korreferent: Prof. Dr. Elke Pogge von Strandmann Table of Contents 1. Summary……………………………………………………………………………...1 1. Zussamenfassung …………………………………………………………....……….2 2. Introduction………………………………………………………………………......3 2.1 The Hedgehog signalling pathway…………………………………………………3 2.2 The Mechanism of Hedgehog signal transduction…………………………………5 2.3 Ptch1 a tumor suppressor gene…………………………………………………....10 2.4 Primary cilium and Hedgehog signalling.………………….………………….….11 2.5 Histone deacetylases (HDACs)………………………………………………...…15 2.6 Classification of Histone deacetylases……………………………………………16 2.7 Histone deacetylase 6……………………………………………………….….…17 2.8 Regulation of Ciliogenesis by HDAC6…………………………………………...20 2.9 Hedgehog signalling and Cancer………………………………………………….22 2.10 Hedgehog signalling Type-I- Mutation driven-ligand independent………….….22 2.11 Hedgehog signalling Type-II-Autocrine-ligand dependent………………….….23 -

Clinical Utility Gene Card For: Joubert Syndrome - Update 2013
European Journal of Human Genetics (2013) 21, doi:10.1038/ejhg.2013.10 & 2013 Macmillan Publishers Limited All rights reserved 1018-4813/13 www.nature.com/ejhg CLINICAL UTILITY GENE CARD UPDATE Clinical utility gene card for: Joubert syndrome - update 2013 Enza Maria Valente*,1,2, Francesco Brancati1, Eugen Boltshauser3 and Bruno Dallapiccola4 European Journal of Human Genetics (2013) 21, doi:10.1038/ejhg.2013.10; published online 13 February 2013 Update to: European Journal of Human Genetics (2011) 19, doi:10.1038/ejhg.2011.49; published online 30 March 2011 1. DISEASE CHARACTERISTICS 1.6 Analytical methods 1.1 Name of the disease (synonyms) Direct sequencing of coding genomic regions and splice site junctions; Joubert syndrome (JS); Joubert-Boltshauser syndrome; Joubert syn- multiplex microsatellite analysis for detection of NPHP1 homozygous drome-related disorders (JSRD), including cerebellar vermis hypo/ deletion. Possibly, qPCR or targeted array-CGH for detection of aplasia, oligophrenia, congenital ataxia, ocular coloboma, and hepatic genomic rearrangements in other genes. fibrosis (COACH) syndrome; cerebellooculorenal, or cerebello-oculo- renal (COR) syndrome; Dekaban-Arima syndrome; Va´radi-Papp 1.7 Analytical validation syndrome or Orofaciodigital type VI (OFDVI) syndrome; Malta Direct sequencing of both DNA strands; verification of sequence and syndrome. qPCR results in an independent experiment. 1.2 OMIM# of the disease 1.8 Estimated frequency of the disease 213300, 243910, 216360, 277170. (incidence at birth-‘birth prevalence’-or population prevalence) No good population-based data on JSRD prevalence have been published. A likely underestimated frequency between 1/80 000 and 1.3 Name of the analysed genes or DNA/chromosome segments 1/100 000 live births is based on unpublished data. -

Abstracts from the 51St European Society of Human Genetics Conference: Electronic Posters
European Journal of Human Genetics (2019) 27:870–1041 https://doi.org/10.1038/s41431-019-0408-3 MEETING ABSTRACTS Abstracts from the 51st European Society of Human Genetics Conference: Electronic Posters © European Society of Human Genetics 2019 June 16–19, 2018, Fiera Milano Congressi, Milan Italy Sponsorship: Publication of this supplement was sponsored by the European Society of Human Genetics. All content was reviewed and approved by the ESHG Scientific Programme Committee, which held full responsibility for the abstract selections. Disclosure Information: In order to help readers form their own judgments of potential bias in published abstracts, authors are asked to declare any competing financial interests. Contributions of up to EUR 10 000.- (Ten thousand Euros, or equivalent value in kind) per year per company are considered "Modest". Contributions above EUR 10 000.- per year are considered "Significant". 1234567890();,: 1234567890();,: E-P01 Reproductive Genetics/Prenatal Genetics then compared this data to de novo cases where research based PO studies were completed (N=57) in NY. E-P01.01 Results: MFSIQ (66.4) for familial deletions was Parent of origin in familial 22q11.2 deletions impacts full statistically lower (p = .01) than for de novo deletions scale intelligence quotient scores (N=399, MFSIQ=76.2). MFSIQ for children with mater- nally inherited deletions (63.7) was statistically lower D. E. McGinn1,2, M. Unolt3,4, T. B. Crowley1, B. S. Emanuel1,5, (p = .03) than for paternally inherited deletions (72.0). As E. H. Zackai1,5, E. Moss1, B. Morrow6, B. Nowakowska7,J. compared with the NY cohort where the MFSIQ for Vermeesch8, A.