An Exome-Wide Sequencing Study of Lipid Response to High-Fat Meal and Fenofibrate in Caucasians from the GOLDN Cohort
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Old Data and Friends Improve with Age: Advancements with the Updated Tools of Genenetwork
bioRxiv preprint doi: https://doi.org/10.1101/2021.05.24.445383; this version posted May 25, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Old data and friends improve with age: Advancements with the updated tools of GeneNetwork Alisha Chunduri1, David G. Ashbrook2 1Department of Biotechnology, Chaitanya Bharathi Institute of Technology, Hyderabad 500075, India 2Department of Genetics, Genomics and Informatics, University of Tennessee Health Science Center, Memphis, TN 38163, USA Abstract Understanding gene-by-environment interactions is important across biology, particularly behaviour. Families of isogenic strains are excellently placed, as the same genome can be tested in multiple environments. The BXD’s recent expansion to 140 strains makes them the largest family of murine isogenic genomes, and therefore give great power to detect QTL. Indefinite reproducible genometypes can be leveraged; old data can be reanalysed with emerging tools to produce novel biological insights. To highlight the importance of reanalyses, we obtained drug- and behavioural-phenotypes from Philip et al. 2010, and reanalysed their data with new genotypes from sequencing, and new models (GEMMA and R/qtl2). We discover QTL on chromosomes 3, 5, 9, 11, and 14, not found in the original study. We narrowed down the candidate genes based on their ability to alter gene expression and/or protein function, using cis-eQTL analysis, and variants predicted to be deleterious. Co-expression analysis (‘gene friends’) and human PheWAS were used to further narrow candidates. -

The Basal Bodies of Chlamydomonas Reinhardtii Susan K
Dutcher and O’Toole Cilia (2016) 5:18 DOI 10.1186/s13630-016-0039-z Cilia REVIEW Open Access The basal bodies of Chlamydomonas reinhardtii Susan K. Dutcher1* and Eileen T. O’Toole2 Abstract The unicellular green alga, Chlamydomonas reinhardtii, is a biflagellated cell that can swim or glide. C. reinhardtii cells are amenable to genetic, biochemical, proteomic, and microscopic analysis of its basal bodies. The basal bodies contain triplet microtubules and a well-ordered transition zone. Both the mother and daughter basal bodies assemble flagella. Many of the proteins found in other basal body-containing organisms are present in the Chlamydomonas genome, and mutants in these genes affect the assembly of basal bodies. Electron microscopic analysis shows that basal body duplication is site-specific and this may be important for the proper duplication and spatial organization of these organelles. Chlamydomonas is an excellent model for the study of basal bodies as well as the transition zone. Keywords: Site-specific basal body duplication, Cartwheel, Transition zone, Centrin fibers Phylogeny and conservation of proteins Centrin, SPD2/CEP192, Asterless/CEP152; CEP70, The green lineage or Viridiplantae consists of the green delta-tubulin, and epsilon-tubulin. Chlamydomonas has algae, which include Chlamydomonas, the angiosperms homologs of all of these based on sequence conservation (the land plants), and the gymnosperms (conifers, cycads, except PLK4, CEP152, and CEP192. Several lines of evi- ginkgos). They are grouped together because they have dence suggests that CEP152, CEP192, and PLK4 interact chlorophyll a and b and lack phycobiliproteins. The green [20, 52] and their concomitant absence in several organ- algae together with the cycads and ginkgos have basal isms suggests that other mechanisms exist that allow for bodies and cilia, while the angiosperms and conifers have control of duplication [4]. -

Par6c Is at the Mother Centriole and Controls Centrosomal Protein
860 Research Article Par6c is at the mother centriole and controls centrosomal protein composition through a Par6a-dependent pathway Vale´rian Dormoy, Kati Tormanen and Christine Su¨ tterlin* Department of Developmental and Cell Biology, University of California, Irvine, Irvine, CA 92697-2300, USA *Author for correspondence ([email protected]) Accepted 3 December 2012 Journal of Cell Science 126, 860–870 ß 2013. Published by The Company of Biologists Ltd doi: 10.1242/jcs.121186 Summary The centrosome contains two centrioles that differ in age, protein composition and function. This non-membrane bound organelle is known to regulate microtubule organization in dividing cells and ciliogenesis in quiescent cells. These specific roles depend on protein appendages at the older, or mother, centriole. In this study, we identified the polarity protein partitioning defective 6 homolog gamma (Par6c) as a novel component of the mother centriole. This specific localization required the Par6c C-terminus, but was independent of intact microtubules, the dynein/dynactin complex and the components of the PAR polarity complex. Par6c depletion resulted in altered centrosomal protein composition, with the loss of a large number of proteins, including Par6a and p150Glued, from the centrosome. As a consequence, there were defects in ciliogenesis, microtubule organization and centrosome reorientation during migration. Par6c interacted with Par3 and aPKC, but these proteins were not required for the regulation of centrosomal protein composition. Par6c also associated with Par6a, which controls protein recruitment to the centrosome through p150Glued. Our study is the first to identify Par6c as a component of the mother centriole and to report a role of a mother centriole protein in the regulation of centrosomal protein composition. -

An Extensive Program of Periodic Alternative Splicing Linked to Cell
RESEARCH ARTICLE An extensive program of periodic alternative splicing linked to cell cycle progression Daniel Dominguez1,2, Yi-Hsuan Tsai1,3, Robert Weatheritt4, Yang Wang1,2, Benjamin J Blencowe4*, Zefeng Wang1,5* 1Department of Pharmacology, University of North Carolina at Chapel Hill, Chapel Hill, United States; 2Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, United States; 3Program in Bioinformatics and Computational Biology, University of North Carolina at Chapel Hill, Chapel Hill, United States; 4Donnelly Centre and Department of Molecular Genetics, University of Toronto, Toronto, Canada; 5Key Lab of Computational Biology, CAS-MPG Partner Institute for Computational Biology, Chinese Academy of Science, Shanghai, China Abstract Progression through the mitotic cell cycle requires periodic regulation of gene function at the levels of transcription, translation, protein-protein interactions, post-translational modification and degradation. However, the role of alternative splicing (AS) in the temporal control of cell cycle is not well understood. By sequencing the human transcriptome through two continuous cell cycles, we identify ~ 1300 genes with cell cycle-dependent AS changes. These genes are significantly enriched in functions linked to cell cycle control, yet they do not significantly overlap genes subject to periodic changes in steady-state transcript levels. Many of the periodically spliced genes are controlled by the SR protein kinase CLK1, whose level undergoes cell cycle- dependent fluctuations via an auto-inhibitory circuit. Disruption of CLK1 causes pleiotropic cell cycle defects and loss of proliferation, whereas CLK1 over-expression is associated with various *For correspondence: cancers. These results thus reveal a large program of CLK1-regulated periodic AS intimately [email protected] (BJB); associated with cell cycle control. -

Molecular Genetics of Microcephaly Primary Hereditary: an Overview
brain sciences Review Molecular Genetics of Microcephaly Primary Hereditary: An Overview Nikistratos Siskos † , Electra Stylianopoulou †, Georgios Skavdis and Maria E. Grigoriou * Department of Molecular Biology & Genetics, Democritus University of Thrace, 68100 Alexandroupolis, Greece; [email protected] (N.S.); [email protected] (E.S.); [email protected] (G.S.) * Correspondence: [email protected] † Equal contribution. Abstract: MicroCephaly Primary Hereditary (MCPH) is a rare congenital neurodevelopmental disorder characterized by a significant reduction of the occipitofrontal head circumference and mild to moderate mental disability. Patients have small brains, though with overall normal architecture; therefore, studying MCPH can reveal not only the pathological mechanisms leading to this condition, but also the mechanisms operating during normal development. MCPH is genetically heterogeneous, with 27 genes listed so far in the Online Mendelian Inheritance in Man (OMIM) database. In this review, we discuss the role of MCPH proteins and delineate the molecular mechanisms and common pathways in which they participate. Keywords: microcephaly; MCPH; MCPH1–MCPH27; molecular genetics; cell cycle 1. Introduction Citation: Siskos, N.; Stylianopoulou, Microcephaly, from the Greek word µικρoκεϕαλi´α (mikrokephalia), meaning small E.; Skavdis, G.; Grigoriou, M.E. head, is a term used to describe a cranium with reduction of the occipitofrontal head circum- Molecular Genetics of Microcephaly ference equal, or more that teo standard deviations -

S41467-019-10037-Y.Pdf
ARTICLE https://doi.org/10.1038/s41467-019-10037-y OPEN Feedback inhibition of cAMP effector signaling by a chaperone-assisted ubiquitin system Laura Rinaldi1, Rossella Delle Donne1, Bruno Catalanotti 2, Omar Torres-Quesada3, Florian Enzler3, Federica Moraca 4, Robert Nisticò5, Francesco Chiuso1, Sonia Piccinin5, Verena Bachmann3, Herbert H Lindner6, Corrado Garbi1, Antonella Scorziello7, Nicola Antonino Russo8, Matthis Synofzik9, Ulrich Stelzl 10, Lucio Annunziato11, Eduard Stefan 3 & Antonio Feliciello1 1234567890():,; Activation of G-protein coupled receptors elevates cAMP levels promoting dissociation of protein kinase A (PKA) holoenzymes and release of catalytic subunits (PKAc). This results in PKAc-mediated phosphorylation of compartmentalized substrates that control central aspects of cell physiology. The mechanism of PKAc activation and signaling have been largely characterized. However, the modes of PKAc inactivation by regulated proteolysis were unknown. Here, we identify a regulatory mechanism that precisely tunes PKAc stability and downstream signaling. Following agonist stimulation, the recruitment of the chaperone- bound E3 ligase CHIP promotes ubiquitylation and proteolysis of PKAc, thus attenuating cAMP signaling. Genetic inactivation of CHIP or pharmacological inhibition of HSP70 enhances PKAc signaling and sustains hippocampal long-term potentiation. Interestingly, primary fibroblasts from autosomal recessive spinocerebellar ataxia 16 (SCAR16) patients carrying germline inactivating mutations of CHIP show a dramatic dysregulation of PKA signaling. This suggests the existence of a negative feedback mechanism for restricting hormonally controlled PKA activities. 1 Department of Molecular Medicine and Medical Biotechnologies, University Federico II, 80131 Naples, Italy. 2 Department of Pharmacy, University Federico II, 80131 Naples, Italy. 3 Institute of Biochemistry and Center for Molecular Biosciences, University of Innsbruck, A-6020 Innsbruck, Austria. -
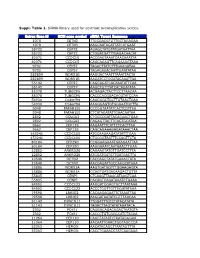
Suppl. Table 1
Suppl. Table 1. SiRNA library used for centriole overduplication screen. Entrez Gene Id NCBI gene symbol siRNA Target Sequence 1070 CETN3 TTGCGACGTGTTGCTAGAGAA 1070 CETN3 AAGCAATAGATTATCATGAAT 55722 CEP72 AGAGCTATGTATGATAATTAA 55722 CEP72 CTGGATGATTTGAGACAACAT 80071 CCDC15 ACCGAGTAAATCAACAAATTA 80071 CCDC15 CAGCAGAGTTCAGAAAGTAAA 9702 CEP57 TAGACTTATCTTTGAAGATAA 9702 CEP57 TAGAGAAACAATTGAATATAA 282809 WDR51B AAGGACTAATTTAAATTACTA 282809 WDR51B AAGATCCTGGATACAAATTAA 55142 CEP27 CAGCAGATCACAAATATTCAA 55142 CEP27 AAGCTGTTTATCACAGATATA 85378 TUBGCP6 ACGAGACTACTTCCTTAACAA 85378 TUBGCP6 CACCCACGGACACGTATCCAA 54930 C14orf94 CAGCGGCTGCTTGTAACTGAA 54930 C14orf94 AAGGGAGTGTGGAAATGCTTA 5048 PAFAH1B1 CCCGGTAATATCACTCGTTAA 5048 PAFAH1B1 CTCATAGATATTGAACAATAA 2802 GOLGA3 CTGGCCGATTACAGAACTGAA 2802 GOLGA3 CAGAGTTACTTCAGTGCATAA 9662 CEP135 AAGAATTTCATTCTCACTTAA 9662 CEP135 CAGCAGAAAGAGATAAACTAA 153241 CCDC100 ATGCAAGAAGATATATTTGAA 153241 CCDC100 CTGCGGTAATTTCCAGTTCTA 80184 CEP290 CCGGAAGAAATGAAGAATTAA 80184 CEP290 AAGGAAATCAATAAACTTGAA 22852 ANKRD26 CAGAAGTATGTTGATCCTTTA 22852 ANKRD26 ATGGATGATGTTGATGACTTA 10540 DCTN2 CACCAGCTATATGAAACTATA 10540 DCTN2 AACGAGATTGCCAAGCATAAA 25886 WDR51A AAGTGATGGTTTGGAAGAGTA 25886 WDR51A CCAGTGATGACAAGACTGTTA 55835 CENPJ CTCAAGTTAAACATAAGTCAA 55835 CENPJ CACAGTCAGATAAATCTGAAA 84902 CCDC123 AAGGATGGAGTGCTTAATAAA 84902 CCDC123 ACCCTGGTTGTTGGATATAAA 79598 LRRIQ2 CACAAGAGAATTCTAAATTAA 79598 LRRIQ2 AAGGATAATATCGTTTAACAA 51143 DYNC1LI1 TTGGATTTGTCTATACATATA 51143 DYNC1LI1 TAGACTTAGTATATAAATACA 2302 FOXJ1 CAGGACAGACAGACTAATGTA -

Association of Cnvs with Methylation Variation
www.nature.com/npjgenmed ARTICLE OPEN Association of CNVs with methylation variation Xinghua Shi1,8, Saranya Radhakrishnan2, Jia Wen1, Jin Yun Chen2, Junjie Chen1,8, Brianna Ashlyn Lam1, Ryan E. Mills 3, ✉ ✉ Barbara E. Stranger4, Charles Lee5,6,7 and Sunita R. Setlur 2 Germline copy number variants (CNVs) and single-nucleotide polymorphisms (SNPs) form the basis of inter-individual genetic variation. Although the phenotypic effects of SNPs have been extensively investigated, the effects of CNVs is relatively less understood. To better characterize mechanisms by which CNVs affect cellular phenotype, we tested their association with variable CpG methylation in a genome-wide manner. Using paired CNV and methylation data from the 1000 genomes and HapMap projects, we identified genome-wide associations by methylation quantitative trait locus (mQTL) analysis. We found individual CNVs being associated with methylation of multiple CpGs and vice versa. CNV-associated methylation changes were correlated with gene expression. CNV-mQTLs were enriched for regulatory regions, transcription factor-binding sites (TFBSs), and were involved in long- range physical interactions with associated CpGs. Some CNV-mQTLs were associated with methylation of imprinted genes. Several CNV-mQTLs and/or associated genes were among those previously reported by genome-wide association studies (GWASs). We demonstrate that germline CNVs in the genome are associated with CpG methylation. Our findings suggest that structural variation together with methylation may affect cellular phenotype. npj Genomic Medicine (2020) 5:41 ; https://doi.org/10.1038/s41525-020-00145-w 1234567890():,; INTRODUCTION influence transcript regulation is DNA methylation, which involves The extent of genetic variation that exists in the human addition of a methyl group to cytosine residues within a CpG population is continually being characterized in efforts to identify dinucleotide. -

Whole Exome Sequencing Identifies Causative Mutations in the Majority of Consanguineous Or Familial Cases with Childhood-Onset I
HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Kidney Manuscript Author Int. Author manuscript; Manuscript Author available in PMC 2016 August 01. Published in final edited form as: Kidney Int. 2016 February ; 89(2): 468–475. doi:10.1038/ki.2015.317. Whole exome sequencing identifies causative mutations in the majority of consanguineous or familial cases with childhood- onset increased renal echogenicity Daniela A. Braun#1, Markus Schueler#1, Jan Halbritter1, Heon Yung Gee1, Jonathan D. Porath1, Jennifer A. Lawson1, Rannar Airik1, Shirlee Shril1, Susan J. Allen2, Deborah Stein1, Adila Al Kindy3, Bodo B. Beck4, Nurcan Cengiz5, Khemchand N. Moorani6, Fatih Ozaltin7,8,9, Seema Hashmi10, John A. Sayer11, Detlef Bockenhauer12, Neveen A. Soliman13,14, Edgar A. Otto2, Richard P. Lifton15,16,17, and Friedhelm Hildebrandt1,17 1Division of Nephrology, Department of Medicine, Boston Children's Hospital, Harvard Medical School, Boston, Massachusetts, USA 2Department of Pediatrics, University of Michigan, Michigan, USA 3Department of Genetics, Sultan Qaboos University Hospital, Sultanate of Oman 4Institute for Human Genetics, University of Cologne, Germany 5Baskent University, School of Medicine, Adana Medical Training and Research Center, Department of Pediatric Nephrology, Adana, Turkey 6Department of Pediatric Nephrology, National Institute of Child Health, Karachi 75510, Pakistan 7Faculty of Medicine, Department of Pediatric Nephrology, Hacettepe University, Ankara, Turkey 8Nephrogenetics Laboratory, Faculty of Medicine, -

TALPID3 and ANKRD26 Selectively Orchestrate FBF1 Localization and Cilia Gating
ARTICLE https://doi.org/10.1038/s41467-020-16042-w OPEN TALPID3 and ANKRD26 selectively orchestrate FBF1 localization and cilia gating Hao Yan 1,2,3,7, Chuan Chen4,7, Huicheng Chen1,2,3, Hui Hong 1, Yan Huang4,5, Kun Ling4,5, ✉ ✉ Jinghua Hu4,5,6 & Qing Wei 3 Transition fibers (TFs) regulate cilia gating and make the primary cilium a distinct functional entity. However, molecular insights into the biogenesis of a functional cilia gate remain 1234567890():,; elusive. In a forward genetic screen in Caenorhabditis elegans, we uncover that TALP-3, a homolog of the Joubert syndrome protein TALPID3, is a TF-associated component. Genetic analysis reveals that TALP-3 coordinates with ANKR-26, the homolog of ANKRD26, to orchestrate proper cilia gating. Mechanistically, TALP-3 and ANKR-26 form a complex with key gating component DYF-19, the homolog of FBF1. Co-depletion of TALP-3 and ANKR-26 specifically impairs the recruitment of DYF-19 to TFs. Interestingly, in mammalian cells, TALPID3 and ANKRD26 also play a conserved role in coordinating the recruitment of FBF1 to TFs. We thus report a conserved protein module that specifically regulates the functional component of the ciliary gate and suggest a correlation between defective gating and cilio- pathy pathogenesis. 1 CAS Key Laboratory of Insect Developmental and Evolutionary Biology, CAS Center for Excellence in Molecular Plant Sciences, Institute of Plant Physiology and Ecology, Chinese Academy of Sciences, Shanghai 200032, China. 2 University of Chinese Academy of Sciences, Beijing 100039, China. 3 Center for Reproduction and Health Development, Institute of Biomedicine and Biotechnology, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences (CAS), Shenzhen 518055, China. -

CEP164-Null Cells Generated by Genome Editing Show a Ciliation Defect with Intact DNA Repair Capacity Owen M
© 2016. Published by The Company of Biologists Ltd | Journal of Cell Science (2016) 129, 1769-1774 doi:10.1242/jcs.186221 SHORT REPORT CEP164-null cells generated by genome editing show a ciliation defect with intact DNA repair capacity Owen M. Daly1, David Gaboriau1,*, Kadin Karakaya2, Sinéad King1, Tiago J. Dantas1,‡, Pierce Lalor3, Peter Dockery3, Alwin Krämer2 and Ciaran G. Morrison1,§ ABSTRACT induced by the removal of growth factors, facilitates ciliogenesis Primary cilia are microtubule structures that extend from the distal end (Kobayashi and Dynlacht, 2011). Current models associate primary of the mature, mother centriole. CEP164 is a component of the distal cilia with cell cycle exit and reduced proliferation, although the appendages carried by the mother centriole that is required for underlying mechanisms of such a link are not well defined (Goto primary cilium formation. Recent data have implicated CEP164 as a et al., 2013). CEP164 ciliopathy gene and suggest that CEP164 plays some roles in the encodes a centriolar appendage protein that is required DNA damage response (DDR). We used reverse genetics to test the for ciliogenesis (Graser et al., 2007; Schmidt et al., 2012). It has also role of CEP164 in the DDR. We found that conditional depletion of been implicated in modulating the DNA damage response (DDR), CEP164 in chicken DT40 cells using an auxin-inducible degron led to particularly CHK1 (Sivasubramaniam et al., 2008). CEP164 was no increase in sensitivity to DNA damage induced by ionising or initially identified in a proteomic analysis of the centrosome and, ultraviolet irradiation. Disruption of CEP164 in human retinal later, as a component of the distal appendages whose depletion by pigmented epithelial cells blocked primary cilium formation but did small interfering RNA (siRNA) treatment caused a marked not affect cellular proliferation or cellular responses to ionising or reduction in primary cilium formation (Andersen et al., 2003; ultraviolet irradiation. -

Cep164 Mediates Vesicular Docking to the Mother Centriole During Early Steps of Ciliogenesis
JCB: Article Cep164 mediates vesicular docking to the mother centriole during early steps of ciliogenesis Kerstin N. Schmidt,1 Stefanie Kuhns,1 Annett Neuner,2 Birgit Hub,3 Hanswalter Zentgraf,3 and Gislene Pereira1 1DKFZ-ZMBH Alliance, German Cancer Research Center, 69120 Heidelberg, Germany 2DKFZ-ZMBH Alliance, Center for Molecular and Cellular Biology, 69120 Heidelberg, Germany 3Department of Tumor Virology, German Cancer Research Center, 69120 Heidelberg, Germany ilia formation is a multi-step process that starts We show that Cep164 was indispensable for the docking with the docking of a vesicle at the distal part of of vesicles at the mother centriole. Using biochemical and C the mother centriole. This step marks the conver- functional assays, we identified the components of the sion of the mother centriole into the basal body, from vesicular transport machinery, the GEF Rabin8 and the which axonemal microtubules extend to form the ciliary GTPase Rab8, as interacting partners of Cep164. We compartment. How vesicles are stably attached to the propose that Cep164 is targeted to the apical domain of mother centriole to initiate ciliary membrane biogenesis the mother centriole to provide the molecular link between is unknown. Here, we investigate the molecular role of the mother centriole and the membrane biogenesis ma- the mother centriolar component Cep164 in ciliogenesis. chinery that initiates cilia formation. Introduction Primary cilia are evolutionarily conserved organelles that play promotes cilia formation and extension via anterograde and retro- an essential role in embryonic development and tissue homeo- grade transport of cargo along the ciliary microtubules (Pedersen stasis in adulthood (D’Angelo and Franco, 2009; Tasouri and and Rosenbaum, 2008).