An Extensive Program of Periodic Alternative Splicing Linked to Cell
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Molecular Genetics of Microcephaly Primary Hereditary: an Overview
brain sciences Review Molecular Genetics of Microcephaly Primary Hereditary: An Overview Nikistratos Siskos † , Electra Stylianopoulou †, Georgios Skavdis and Maria E. Grigoriou * Department of Molecular Biology & Genetics, Democritus University of Thrace, 68100 Alexandroupolis, Greece; [email protected] (N.S.); [email protected] (E.S.); [email protected] (G.S.) * Correspondence: [email protected] † Equal contribution. Abstract: MicroCephaly Primary Hereditary (MCPH) is a rare congenital neurodevelopmental disorder characterized by a significant reduction of the occipitofrontal head circumference and mild to moderate mental disability. Patients have small brains, though with overall normal architecture; therefore, studying MCPH can reveal not only the pathological mechanisms leading to this condition, but also the mechanisms operating during normal development. MCPH is genetically heterogeneous, with 27 genes listed so far in the Online Mendelian Inheritance in Man (OMIM) database. In this review, we discuss the role of MCPH proteins and delineate the molecular mechanisms and common pathways in which they participate. Keywords: microcephaly; MCPH; MCPH1–MCPH27; molecular genetics; cell cycle 1. Introduction Citation: Siskos, N.; Stylianopoulou, Microcephaly, from the Greek word µικρoκεϕαλi´α (mikrokephalia), meaning small E.; Skavdis, G.; Grigoriou, M.E. head, is a term used to describe a cranium with reduction of the occipitofrontal head circum- Molecular Genetics of Microcephaly ference equal, or more that teo standard deviations -

S41467-019-10037-Y.Pdf
ARTICLE https://doi.org/10.1038/s41467-019-10037-y OPEN Feedback inhibition of cAMP effector signaling by a chaperone-assisted ubiquitin system Laura Rinaldi1, Rossella Delle Donne1, Bruno Catalanotti 2, Omar Torres-Quesada3, Florian Enzler3, Federica Moraca 4, Robert Nisticò5, Francesco Chiuso1, Sonia Piccinin5, Verena Bachmann3, Herbert H Lindner6, Corrado Garbi1, Antonella Scorziello7, Nicola Antonino Russo8, Matthis Synofzik9, Ulrich Stelzl 10, Lucio Annunziato11, Eduard Stefan 3 & Antonio Feliciello1 1234567890():,; Activation of G-protein coupled receptors elevates cAMP levels promoting dissociation of protein kinase A (PKA) holoenzymes and release of catalytic subunits (PKAc). This results in PKAc-mediated phosphorylation of compartmentalized substrates that control central aspects of cell physiology. The mechanism of PKAc activation and signaling have been largely characterized. However, the modes of PKAc inactivation by regulated proteolysis were unknown. Here, we identify a regulatory mechanism that precisely tunes PKAc stability and downstream signaling. Following agonist stimulation, the recruitment of the chaperone- bound E3 ligase CHIP promotes ubiquitylation and proteolysis of PKAc, thus attenuating cAMP signaling. Genetic inactivation of CHIP or pharmacological inhibition of HSP70 enhances PKAc signaling and sustains hippocampal long-term potentiation. Interestingly, primary fibroblasts from autosomal recessive spinocerebellar ataxia 16 (SCAR16) patients carrying germline inactivating mutations of CHIP show a dramatic dysregulation of PKA signaling. This suggests the existence of a negative feedback mechanism for restricting hormonally controlled PKA activities. 1 Department of Molecular Medicine and Medical Biotechnologies, University Federico II, 80131 Naples, Italy. 2 Department of Pharmacy, University Federico II, 80131 Naples, Italy. 3 Institute of Biochemistry and Center for Molecular Biosciences, University of Innsbruck, A-6020 Innsbruck, Austria. -
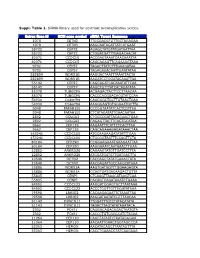
Suppl. Table 1
Suppl. Table 1. SiRNA library used for centriole overduplication screen. Entrez Gene Id NCBI gene symbol siRNA Target Sequence 1070 CETN3 TTGCGACGTGTTGCTAGAGAA 1070 CETN3 AAGCAATAGATTATCATGAAT 55722 CEP72 AGAGCTATGTATGATAATTAA 55722 CEP72 CTGGATGATTTGAGACAACAT 80071 CCDC15 ACCGAGTAAATCAACAAATTA 80071 CCDC15 CAGCAGAGTTCAGAAAGTAAA 9702 CEP57 TAGACTTATCTTTGAAGATAA 9702 CEP57 TAGAGAAACAATTGAATATAA 282809 WDR51B AAGGACTAATTTAAATTACTA 282809 WDR51B AAGATCCTGGATACAAATTAA 55142 CEP27 CAGCAGATCACAAATATTCAA 55142 CEP27 AAGCTGTTTATCACAGATATA 85378 TUBGCP6 ACGAGACTACTTCCTTAACAA 85378 TUBGCP6 CACCCACGGACACGTATCCAA 54930 C14orf94 CAGCGGCTGCTTGTAACTGAA 54930 C14orf94 AAGGGAGTGTGGAAATGCTTA 5048 PAFAH1B1 CCCGGTAATATCACTCGTTAA 5048 PAFAH1B1 CTCATAGATATTGAACAATAA 2802 GOLGA3 CTGGCCGATTACAGAACTGAA 2802 GOLGA3 CAGAGTTACTTCAGTGCATAA 9662 CEP135 AAGAATTTCATTCTCACTTAA 9662 CEP135 CAGCAGAAAGAGATAAACTAA 153241 CCDC100 ATGCAAGAAGATATATTTGAA 153241 CCDC100 CTGCGGTAATTTCCAGTTCTA 80184 CEP290 CCGGAAGAAATGAAGAATTAA 80184 CEP290 AAGGAAATCAATAAACTTGAA 22852 ANKRD26 CAGAAGTATGTTGATCCTTTA 22852 ANKRD26 ATGGATGATGTTGATGACTTA 10540 DCTN2 CACCAGCTATATGAAACTATA 10540 DCTN2 AACGAGATTGCCAAGCATAAA 25886 WDR51A AAGTGATGGTTTGGAAGAGTA 25886 WDR51A CCAGTGATGACAAGACTGTTA 55835 CENPJ CTCAAGTTAAACATAAGTCAA 55835 CENPJ CACAGTCAGATAAATCTGAAA 84902 CCDC123 AAGGATGGAGTGCTTAATAAA 84902 CCDC123 ACCCTGGTTGTTGGATATAAA 79598 LRRIQ2 CACAAGAGAATTCTAAATTAA 79598 LRRIQ2 AAGGATAATATCGTTTAACAA 51143 DYNC1LI1 TTGGATTTGTCTATACATATA 51143 DYNC1LI1 TAGACTTAGTATATAAATACA 2302 FOXJ1 CAGGACAGACAGACTAATGTA -

Association of Cnvs with Methylation Variation
www.nature.com/npjgenmed ARTICLE OPEN Association of CNVs with methylation variation Xinghua Shi1,8, Saranya Radhakrishnan2, Jia Wen1, Jin Yun Chen2, Junjie Chen1,8, Brianna Ashlyn Lam1, Ryan E. Mills 3, ✉ ✉ Barbara E. Stranger4, Charles Lee5,6,7 and Sunita R. Setlur 2 Germline copy number variants (CNVs) and single-nucleotide polymorphisms (SNPs) form the basis of inter-individual genetic variation. Although the phenotypic effects of SNPs have been extensively investigated, the effects of CNVs is relatively less understood. To better characterize mechanisms by which CNVs affect cellular phenotype, we tested their association with variable CpG methylation in a genome-wide manner. Using paired CNV and methylation data from the 1000 genomes and HapMap projects, we identified genome-wide associations by methylation quantitative trait locus (mQTL) analysis. We found individual CNVs being associated with methylation of multiple CpGs and vice versa. CNV-associated methylation changes were correlated with gene expression. CNV-mQTLs were enriched for regulatory regions, transcription factor-binding sites (TFBSs), and were involved in long- range physical interactions with associated CpGs. Some CNV-mQTLs were associated with methylation of imprinted genes. Several CNV-mQTLs and/or associated genes were among those previously reported by genome-wide association studies (GWASs). We demonstrate that germline CNVs in the genome are associated with CpG methylation. Our findings suggest that structural variation together with methylation may affect cellular phenotype. npj Genomic Medicine (2020) 5:41 ; https://doi.org/10.1038/s41525-020-00145-w 1234567890():,; INTRODUCTION influence transcript regulation is DNA methylation, which involves The extent of genetic variation that exists in the human addition of a methyl group to cytosine residues within a CpG population is continually being characterized in efforts to identify dinucleotide. -

An Exome-Wide Sequencing Study of Lipid Response to High-Fat Meal and Fenofibrate in Caucasians from the GOLDN Cohort
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2018 An exome-wide sequencing study of lipid response to high-fat meal and fenofibrate in Caucasians from the GOLDN cohort Xin Geng Ping An Mary F. Feitosa Michael A. Province et al. Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Supplemental Material can be found at: http://www.jlr.org/content/suppl/2018/02/20/jlr.P080333.DC1 .html patient-oriented and epidemiological research An exome-wide sequencing study of lipid response to high-fat meal and fenofibrate in Caucasians from the GOLDN cohort Xin Geng,* Marguerite R. Irvin,† Bertha Hidalgo,† Stella Aslibekyan,† Vinodh Srinivasasainagendra,§ Ping An,‡ Alexis C. Frazier-Wood,|| Hemant K. Tiwari,§ Tushar Dave,# Kathleen Ryan,# Jose M. Ordovas,$,**,†† Robert J. Straka,§§ Mary F. Feitosa,‡ Paul N. Hopkins,‡‡ Ingrid Borecki,|| || Michael A. Province,‡ Braxton D. Mitchell,# Donna K. Arnett,1,## and Degui Zhi1,*,$$ School of Biomedical Informatics* and School of Public Health,$$ The University of Texas Health Downloaded from Science Center at Houston, Houston, TX 77030; Departments of Epidemiology† and Biostatistics,§ University of Alabama at Birmingham, Birmingham, AL 35233; Division of Statistical Genomics, Department of Genetics,‡ Washington University School of Medicine, St. Louis, MO 63110; US Department of Agriculture/Agricultural Research Service Children’s Nutrition Research Center,|| Baylor College of Medicine, Houston, TX 77030; Department of Medicine, Division -

Network-Based Prediction of Drug Combinations
Corrected: Publisher correction ARTICLE https://doi.org/10.1038/s41467-019-09186-x OPEN Network-based prediction of drug combinations Feixiong Cheng1,2,3,4,5, Istvań A. Kovacś1,2 & Albert-Laszló ́Barabasí1,2,6,7 Drug combinations, offering increased therapeutic efficacy and reduced toxicity, play an important role in treating multiple complex diseases. Yet, our ability to identify and validate effective combinations is limited by a combinatorial explosion, driven by both the large number of drug pairs as well as dosage combinations. Here we propose a network-based methodology to identify clinically efficacious drug combinations for specific diseases. By 1234567890():,; quantifying the network-based relationship between drug targets and disease proteins in the human protein–protein interactome, we show the existence of six distinct classes of drug–drug–disease combinations. Relying on approved drug combinations for hypertension and cancer, we find that only one of the six classes correlates with therapeutic effects: if the targets of the drugs both hit disease module, but target separate neighborhoods. This finding allows us to identify and validate antihypertensive combinations, offering a generic, powerful network methodology to identify efficacious combination therapies in drug development. 1 Center for Complex Networks Research and Department of Physics, Northeastern University, Boston, MA 02115, USA. 2 Center for Cancer Systems Biology and Department of Cancer Biology, Dana-Farber Cancer Institute, Boston, MA 02215, USA. 3 Genomic Medicine Institute, Lerner Research Institute, Cleveland Clinic, Cleveland, OH 44106, USA. 4 Department of Molecular Medicine, Cleveland Clinic Lerner College of Medicine, Case Western Reserve University, Cleveland, OH 44195, USA. 5 Case Comprehensive Cancer Center, Case Western Reserve University School of Medicine, Cleveland, OH 44106, USA. -

Downregulation of Carnitine Acyl-Carnitine Translocase by Mirnas
Page 1 of 288 Diabetes 1 Downregulation of Carnitine acyl-carnitine translocase by miRNAs 132 and 212 amplifies glucose-stimulated insulin secretion Mufaddal S. Soni1, Mary E. Rabaglia1, Sushant Bhatnagar1, Jin Shang2, Olga Ilkayeva3, Randall Mynatt4, Yun-Ping Zhou2, Eric E. Schadt6, Nancy A.Thornberry2, Deborah M. Muoio5, Mark P. Keller1 and Alan D. Attie1 From the 1Department of Biochemistry, University of Wisconsin, Madison, Wisconsin; 2Department of Metabolic Disorders-Diabetes, Merck Research Laboratories, Rahway, New Jersey; 3Sarah W. Stedman Nutrition and Metabolism Center, Duke Institute of Molecular Physiology, 5Departments of Medicine and Pharmacology and Cancer Biology, Durham, North Carolina. 4Pennington Biomedical Research Center, Louisiana State University system, Baton Rouge, Louisiana; 6Institute for Genomics and Multiscale Biology, Mount Sinai School of Medicine, New York, New York. Corresponding author Alan D. Attie, 543A Biochemistry Addition, 433 Babcock Drive, Department of Biochemistry, University of Wisconsin-Madison, Madison, Wisconsin, (608) 262-1372 (Ph), (608) 263-9608 (fax), [email protected]. Running Title: Fatty acyl-carnitines enhance insulin secretion Abstract word count: 163 Main text Word count: 3960 Number of tables: 0 Number of figures: 5 Diabetes Publish Ahead of Print, published online June 26, 2014 Diabetes Page 2 of 288 2 ABSTRACT We previously demonstrated that micro-RNAs 132 and 212 are differentially upregulated in response to obesity in two mouse strains that differ in their susceptibility to obesity-induced diabetes. Here we show the overexpression of micro-RNAs 132 and 212 enhances insulin secretion (IS) in response to glucose and other secretagogues including non-fuel stimuli. We determined that carnitine acyl-carnitine translocase (CACT, Slc25a20) is a direct target of these miRNAs. -

Plk4-Induced Centriole Biogenesis in Human Cells
Plk4-induced Centriole Biogenesis in Human Cells Dissertation zur Erlangung des Doktorgrades der Naturwissenschaften der Fakultät für Biologie der Ludwig-Maximilians Universität München Vorgelegt von Julia Kleylein-Sohn München, 2007 Dissertation eingereicht am: 27.11.2007 Tag der mündlichen Prüfung: 18.04.2008 Erstgutachter: Prof. E. A. Nigg Zweitgutachter: PD Dr. Angelika Böttger 2 Hiermit erkläre ich, dass ich die vorliegende Dissertation selbständig und ohne unerlaubte Hilfe angefertigt habe. Sämtliche Experimente wurden von mir selbst durchgeführt, soweit nicht explizit auf Dritte verwiesen wird. Ich habe weder an anderer Stelle versucht, eine Dissertation oder Teile einer solchen einzureichen bzw. einer Prüfungskommission vorzulegen, noch eine Doktorprüfung zu absolvieren. München, den 22.11.2007 3 TABLE OF CONTENTS SUMMARY…………………………………………………………………………..………. 6 INTRODUCTION……………………………………………………………………………. 7 Structure of the centrosome…………………………………………………………….. 7 The centrosome cycle…………………………………………………………………..10 Kinases involved in the regulation of centriole duplication………………………….12 Maintenance of centrosome numbers………………………………………………...13 Licensing of centriole duplication……………………………………………………... 15 ‘De novo ’ centriole assembly pathways in mammalian cells…………………..…...15 Templated centriole biogenesis in mammalian cells……………………………….. 18 The role of centrins and Sfi1p in centrosome duplication ……………………...…..19 Centriole biogenesis in C. elegans …………………………………………………… 21 Centriole biogenesis in human cells………………………………………………….. 23 Centrosome -

A Dissertation Entitled JAK2 Tyrosine Kinase Phosphorylates and Is
A Dissertation Entitled JAK2 Tyrosine Kinase Phosphorylates and is Negatively Regulated by the Centrosomal Protein Ninein By Jennifer Jay Submitted to the Graduate Faculty as partial fulfillment of the requirements for the Doctor of Philosophy Degree in Biology _________________________________________ Dr. Maria Diakonova, Committee Chair _________________________________________ Dr. Patricia R. Komuniecki, Dean College of Graduate Studies The University of Toledo May 2015 Copyright 2015, Jennifer Jay This document is copyrighted material. Under copyright law, no parts of this document may be reproduced without the expressed permission of the author. An Abstract of JAK2 Tyrosine Kinase Phosphorylates and is Negatively Regulated by the Centrosomal Protein Ninein By Jennifer Jay Submitted to the Graduate Faculty as partial fulfillment of the requirements for the Doctor of Philosophy Degree in Biology The University of Toledo May 2015 Janus Kinase 2 (JAK2) is a non-receptor tyrosine kinase that is activated by two- thirds of the cytokine receptor superfamily including receptors to interferon-γ, growth hormone and prolactin. Upon ligand binding to its receptor, JAK2 becomes activated and can phosphorylate downstream targets that lead to diverse physiological responses. Even though JAK2 plays an important role in cytokine signaling, its subcellular localization is still under debate. We show that both inactive and active JAK2 (pJAK2) localizes around the mother centrioles where it partially colocalizes with ninein, a protein involved in microtubule (MT) nucleation and anchoring. We demonstrate that depletion of JAK2 or using JAK2-null cells, results in microtubule anchorage defects at the mother centriole and an increased number of cells with mitotic defects; however, MT nucleation is unaffected. -
![Ordered Layers and Scaffolding Gels[Version 1; Referees: 3 Approved]](https://docslib.b-cdn.net/cover/4312/ordered-layers-and-scaffolding-gels-version-1-referees-3-approved-2644312.webp)
Ordered Layers and Scaffolding Gels[Version 1; Referees: 3 Approved]
F1000Research 2017, 6(F1000 Faculty Rev):1622 Last updated: 31 AUG 2017 REVIEW Recent advances in pericentriolar material organization: ordered layers and scaffolding gels [version 1; referees: 3 approved] Andrew M. Fry , Josephina Sampson, Caroline Shak, Sue Shackleton Department of Molecular and Cell Biology, University of Leicester, Leicester, UK First published: 31 Aug 2017, 6(F1000 Faculty Rev):1622 (doi: Open Peer Review v1 10.12688/f1000research.11652.1) Latest published: 31 Aug 2017, 6(F1000 Faculty Rev):1622 (doi: 10.12688/f1000research.11652.1) Referee Status: Abstract Invited Referees The centrosome is an unusual organelle that lacks a surrounding membrane, 1 2 3 raising the question of what limits its size and shape. Moreover, while electron microscopy (EM) has provided a detailed view of centriole architecture, there version 1 has been limited understanding of how the second major component of published centrosomes, the pericentriolar material (PCM), is organized. Here, we 31 Aug 2017 summarize exciting recent findings from super-resolution fluorescence imaging, structural biology, and biochemical reconstitution that together reveal the presence of ordered layers and complex gel-like scaffolds in the PCM. F1000 Faculty Reviews are commissioned Moreover, we discuss how this is leading to a better understanding of the from members of the prestigious F1000 process of microtubule nucleation, how alterations in PCM size are regulated in Faculty. In order to make these reviews as cycling and differentiated cells, and why mutations in PCM components lead to comprehensive and accessible as possible, specific human pathologies. peer review takes place before publication; the referees are listed below, but their reports are not formally published. -

A SARS-Cov-2 Protein Interaction Map Reveals Targets for Drug Repurposing
Article A SARS-CoV-2 protein interaction map reveals targets for drug repurposing https://doi.org/10.1038/s41586-020-2286-9 A list of authors and affiliations appears at the end of the paper Received: 23 March 2020 Accepted: 22 April 2020 A newly described coronavirus named severe acute respiratory syndrome Published online: 30 April 2020 coronavirus 2 (SARS-CoV-2), which is the causative agent of coronavirus disease 2019 (COVID-19), has infected over 2.3 million people, led to the death of more than Check for updates 160,000 individuals and caused worldwide social and economic disruption1,2. There are no antiviral drugs with proven clinical efcacy for the treatment of COVID-19, nor are there any vaccines that prevent infection with SARS-CoV-2, and eforts to develop drugs and vaccines are hampered by the limited knowledge of the molecular details of how SARS-CoV-2 infects cells. Here we cloned, tagged and expressed 26 of the 29 SARS-CoV-2 proteins in human cells and identifed the human proteins that physically associated with each of the SARS-CoV-2 proteins using afnity-purifcation mass spectrometry, identifying 332 high-confdence protein–protein interactions between SARS-CoV-2 and human proteins. Among these, we identify 66 druggable human proteins or host factors targeted by 69 compounds (of which, 29 drugs are approved by the US Food and Drug Administration, 12 are in clinical trials and 28 are preclinical compounds). We screened a subset of these in multiple viral assays and found two sets of pharmacological agents that displayed antiviral activity: inhibitors of mRNA translation and predicted regulators of the sigma-1 and sigma-2 receptors. -

Metabolism, Ubiquinone Synthesis, and Longevity
Downloaded from genesdev.cshlp.org on September 25, 2021 - Published by Cold Spring Harbor Laboratory Press PERSPECTIVE Metabolism, ubiquinone synthesis, and longevity Hugo Aguilaniu,1 Jenni Durieux,1 and Andrew Dillin2 Molecular and Cell Biology Laboratory, The Salk Institute for Biological Studies, La Jolla, California 92037, USA “You have made your way from worm to man, but much gram of tissue and lived longer than animals with higher within you is still a worm.” metabolic rates. Exceptions have been discovered since —Nieztsche Pearl’s initial observations, such as the high metabolic rates of some long-lived birds (Holmes et al. 2001); how- Until recently, the mechanisms that control the aging ever, Pearl’s initial observation led to the formulation of process were thought to be immensely complex and the “rate of living theory of aging” (Pearl 1928). The nearly impossible to dissect at the molecular level. How- theory suggests that reduced metabolic rates in an ani- ever, genetic analysis, primarily in model organisms mal should result in an increased life span. such as yeast, worms, and flies, is dramatically changing Several years later, the “rate of living theory of aging” this view. It seems that at least three distinct genetic became more refined with the formulation of the “oxy- networks control the aging process. These networks in- gen radical theory of aging” proposed by Denham Har- clude the insulin/IGF-1 signaling pathway, signaling man (Harman 1956). Harman reasoned that reactions us- that arises from the mitochondrial electron transport • ing molecular oxygen create, on occasion, toxic O − radi- chain, and finally, mechanisms that govern the response 2 cals.