Genetic Basis of Lactation and Lactation Efficiency in Pigs Dinesh Moorkattukara Thekkoot Iowa State University
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Chromosome Level Genome of Astyanax Mexicanus Surface Fish for Comparing Population
bioRxiv preprint doi: https://doi.org/10.1101/2020.07.06.189654; this version posted July 6, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Title 2 A chromosome level genome of Astyanax mexicanus surface fish for comparing population- 3 specific genetic differences contributing to trait evolution. 4 5 Authors 6 Wesley C. Warren1, Tyler E. Boggs2, Richard Borowsky3, Brian M. Carlson4, Estephany 7 Ferrufino5, Joshua B. Gross2, LaDeana Hillier6, Zhilian Hu7, Alex C. Keene8, Alexander Kenzior9, 8 Johanna E. Kowalko5, Chad Tomlinson10, Milinn Kremitzki10, Madeleine E. Lemieux11, Tina 9 Graves-Lindsay10, Suzanne E. McGaugh12, Jeff T. Miller12, Mathilda Mommersteeg7, Rachel L. 10 Moran12, Robert Peuß9, Edward Rice1, Misty R. Riddle13, Itzel Sifuentes-Romero5, Bethany A. 11 Stanhope5,8, Clifford J. Tabin13, Sunishka Thakur5, Yamamoto Yoshiyuki14, Nicolas Rohner9,15 12 13 Authors for correspondence: Wesley C. Warren ([email protected]), Nicolas Rohner 14 ([email protected]) 15 16 Affiliation 17 1Department of Animal Sciences, Department of Surgery, Institute for Data Science and 18 Informatics, University of Missouri, Bond Life Sciences Center, Columbia, MO 19 2 Department of Biological Sciences, University of Cincinnati, Cincinnati, OH 20 3 Department of Biology, New York University, New York, NY 21 4 Department of Biology, The College of Wooster, Wooster, OH 22 5 Harriet L. Wilkes Honors College, Florida Atlantic University, Jupiter FL 23 6 Department of Genome Sciences, University of Washington, Seattle, WA 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.07.06.189654; this version posted July 6, 2020. -

Gene Expression Profile of THP-1 Cells Treated with Heat-Killed Candida Albicans
Original Article Page 1 of 5 Gene expression profile of THP-1 cells treated with heat-killed Candida albicans Zhi-De Hu1,2*, Ting-Ting Wei1*, Qing-Qin Tang1, Ning Ma1, Li-Li Wang1, Bao-Dong Qin1, Jian-Rong Yin1, Lin Zhou1, Ren-Qian Zhong1 1Department of Laboratory Medicine, Changzheng Hospital, Second Military Medical University, Shanghai 200003, China; 2Department of Laboratory Medicine, General Hospital, Ji’nan Military Region of PLA, Ji’nan 250031, China Contributions: (I) Conception and design: RQ Zhong, L Zhou, ZD Hu; (II) Administrative support: RQ Zhong; (III) Provision of study materials or patients: All authors; (IV) Collection and assembly of data: All authors; (V) Data analysis and interpretation: All authors; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. *These authors contributed equally to this work. Correspondence to: Ren-Qian Zhong, MD, PhD. Department of Laboratory Medicine, Changzheng Hospital, the Second Military Medical University, Shanghai 200003, China. Email: [email protected]; Lin Zhou, MD, PhD. Department of Laboratory Medicine, Changzheng Hospital, the Second Military Medical University, Shanghai 200003, China. Email: [email protected]. Background: Mechanisms under immune response against Candida albicans (C. albicans) remain largely unknown. To better understand the mechanisms of innate immune response against C. albicans, we analyzed the gene expression profile of THP-1 cells stimulated with heat-killed C. albicans. Methods: THP-1 cells were stimulated with heat-killed C. albicans for 9 hours at a ratio of 1:1, and gene expression profile of the cells was analyzed using Whole Human Genome Oligo Microarray. Differentially expressed genes were defined as change folds more than 2 and with statistical significance. -

Nº Ref Uniprot Proteína Péptidos Identificados Por MS/MS 1 P01024
Document downloaded from http://www.elsevier.es, day 26/09/2021. This copy is for personal use. Any transmission of this document by any media or format is strictly prohibited. Nº Ref Uniprot Proteína Péptidos identificados 1 P01024 CO3_HUMAN Complement C3 OS=Homo sapiens GN=C3 PE=1 SV=2 por 162MS/MS 2 P02751 FINC_HUMAN Fibronectin OS=Homo sapiens GN=FN1 PE=1 SV=4 131 3 P01023 A2MG_HUMAN Alpha-2-macroglobulin OS=Homo sapiens GN=A2M PE=1 SV=3 128 4 P0C0L4 CO4A_HUMAN Complement C4-A OS=Homo sapiens GN=C4A PE=1 SV=1 95 5 P04275 VWF_HUMAN von Willebrand factor OS=Homo sapiens GN=VWF PE=1 SV=4 81 6 P02675 FIBB_HUMAN Fibrinogen beta chain OS=Homo sapiens GN=FGB PE=1 SV=2 78 7 P01031 CO5_HUMAN Complement C5 OS=Homo sapiens GN=C5 PE=1 SV=4 66 8 P02768 ALBU_HUMAN Serum albumin OS=Homo sapiens GN=ALB PE=1 SV=2 66 9 P00450 CERU_HUMAN Ceruloplasmin OS=Homo sapiens GN=CP PE=1 SV=1 64 10 P02671 FIBA_HUMAN Fibrinogen alpha chain OS=Homo sapiens GN=FGA PE=1 SV=2 58 11 P08603 CFAH_HUMAN Complement factor H OS=Homo sapiens GN=CFH PE=1 SV=4 56 12 P02787 TRFE_HUMAN Serotransferrin OS=Homo sapiens GN=TF PE=1 SV=3 54 13 P00747 PLMN_HUMAN Plasminogen OS=Homo sapiens GN=PLG PE=1 SV=2 48 14 P02679 FIBG_HUMAN Fibrinogen gamma chain OS=Homo sapiens GN=FGG PE=1 SV=3 47 15 P01871 IGHM_HUMAN Ig mu chain C region OS=Homo sapiens GN=IGHM PE=1 SV=3 41 16 P04003 C4BPA_HUMAN C4b-binding protein alpha chain OS=Homo sapiens GN=C4BPA PE=1 SV=2 37 17 Q9Y6R7 FCGBP_HUMAN IgGFc-binding protein OS=Homo sapiens GN=FCGBP PE=1 SV=3 30 18 O43866 CD5L_HUMAN CD5 antigen-like OS=Homo -

Novel Signature Genes for Human Left Ventricle Cardiomyopathies Identifed by Weighted Co- Expression Network Analysis (WGCNA)
Novel Signature Genes for Human Left Ventricle Cardiomyopathies Identied by Weighted Co- expression Network Analysis (WGCNA) Jiao Tian Kunming Institute of Botany Chinese Academy of Sciences Zhengyuan Wu Kunming Institute of Botany Chinese Academy of Sciences Yaqi Zhang Kunming Institute of Botany Chinese Academy of Sciences Yingying He Yunnan University SHUBAI LIU ( [email protected] ) Kunming Institute of Botany Chinese Academy of Sciences Research Keywords: Heart failure, Cardiomyopathy, Ventricle cardiomyocytes, WGCNA, Signature gene Posted Date: October 8th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-86136/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/29 Abstract Background Cardiomyopathy, a heart disease that arises from different etiologies, brings a huge healthcare burden to the global society. Identication of biomarkers can be very useful for early diagnosis of cardiomyopathy, interruption of the disease procession to heart failure, and decrement of the mortality. Methods Clinical cases of cardiomyopathy were screened out of independently investigations from the genomic database. Exploration of its whole transcription disorder pattern by WGCNA (Weighted Gene Co- expression Network Analysis) to discover the signature genes for different subtypes of cardiomyopathy. The hub genes and key pathways, which are correlated to cardiomyopathy traits, were identied through co-expression and protein-protein interaction (PPI) networks enrichment analysis. Discovered hub genes were blast through the Cardiovascular Disease Portal to verify functions related to human cardiomyopathies. Results Three common axes of signature genes were discovered for ve subtypes of cardiomyopathy: 1) Four genes (MDM4, CFLAR, RPS6KB1, PKD1L2) were common for ischemic and ischemic cardiomyopathy subgroups; 2) Subtypes of cardiomyopathy (ischemic, post. -

Supplementary Tables S1-S3
Supplementary Table S1: Real time RT-PCR primers COX-2 Forward 5’- CCACTTCAAGGGAGTCTGGA -3’ Reverse 5’- AAGGGCCCTGGTGTAGTAGG -3’ Wnt5a Forward 5’- TGAATAACCCTGTTCAGATGTCA -3’ Reverse 5’- TGTACTGCATGTGGTCCTGA -3’ Spp1 Forward 5'- GACCCATCTCAGAAGCAGAA -3' Reverse 5'- TTCGTCAGATTCATCCGAGT -3' CUGBP2 Forward 5’- ATGCAACAGCTCAACACTGC -3’ Reverse 5’- CAGCGTTGCCAGATTCTGTA -3’ Supplementary Table S2: Genes synergistically regulated by oncogenic Ras and TGF-β AU-rich probe_id Gene Name Gene Symbol element Fold change RasV12 + TGF-β RasV12 TGF-β 1368519_at serine (or cysteine) peptidase inhibitor, clade E, member 1 Serpine1 ARE 42.22 5.53 75.28 1373000_at sushi-repeat-containing protein, X-linked 2 (predicted) Srpx2 19.24 25.59 73.63 1383486_at Transcribed locus --- ARE 5.93 27.94 52.85 1367581_a_at secreted phosphoprotein 1 Spp1 2.46 19.28 49.76 1368359_a_at VGF nerve growth factor inducible Vgf 3.11 4.61 48.10 1392618_at Transcribed locus --- ARE 3.48 24.30 45.76 1398302_at prolactin-like protein F Prlpf ARE 1.39 3.29 45.23 1392264_s_at serine (or cysteine) peptidase inhibitor, clade E, member 1 Serpine1 ARE 24.92 3.67 40.09 1391022_at laminin, beta 3 Lamb3 2.13 3.31 38.15 1384605_at Transcribed locus --- 2.94 14.57 37.91 1367973_at chemokine (C-C motif) ligand 2 Ccl2 ARE 5.47 17.28 37.90 1369249_at progressive ankylosis homolog (mouse) Ank ARE 3.12 8.33 33.58 1398479_at ryanodine receptor 3 Ryr3 ARE 1.42 9.28 29.65 1371194_at tumor necrosis factor alpha induced protein 6 Tnfaip6 ARE 2.95 7.90 29.24 1386344_at Progressive ankylosis homolog (mouse) -

The Consensus Coding Sequences of Human Breast and Colorectal Cancers Tobias Sjöblom,1* Siân Jones,1* Laura D
The Consensus Coding Sequences of Human Breast and Colorectal Cancers Tobias Sjöblom,1* Siân Jones,1* Laura D. Wood,1* D. Williams Parsons,1* Jimmy Lin,1 Thomas Barber,1 Diana Mandelker,1 Rebecca J. Leary,1 Janine Ptak,1 Natalie Silliman,1 Steve Szabo,1 Phillip Buckhaults,2 Christopher Farrell,2 Paul Meeh,2 Sanford D. Markowitz,3 Joseph Willis,4 Dawn Dawson,4 James K. V. Willson,5 Adi F. Gazdar,6 James Hartigan,7 Leo Wu,8 Changsheng Liu,8 Giovanni Parmigiani,9 Ben Ho Park,10 Kurtis E. Bachman,11 Nickolas Papadopoulos,1 Bert Vogelstein,1† Kenneth W. Kinzler,1† Victor E. Velculescu1† 1Ludwig Center and Howard Hughes Medical Institute, Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins, Baltimore, MD 21231, USA. 2Department of Pathology and Microbiology, Center for Colon Cancer Research, and South Carolina Cancer Center, Division of Basic Research, University of South Carolina School of Medicine, Columbia, SC 29229, USA. 3Department of Medicine, Ireland Cancer Center, and Howard Hughes Medical Institute, Case Western Reserve University and University Hospitals of Cleveland, Cleveland, OH 44106, USA. 4Department of Pathology and Ireland Cancer Center, Case Western Reserve University and University Hospitals of Cleveland, Cleveland, OH 44106, USA. 5Harold C. Simmons Comprehensive Cancer Center, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA. 6Hamon Center for Therapeutic Oncology Research and Department of Pathology, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA. 7Agencourt Bioscience Corporation, Beverly, MA 01915, USA. 8SoftGenetics LLC, State College, PA 16803, USA. 9Departments of Oncology, Biostatistics, and Pathology, Johns Hopkins Medical Institutions, Baltimore, MD 21205, USA. -

Identification of Gene-Oriented Exon Orthology Between Human and Mouse
Fu and Lin BMC Genomics 2012, 13(Suppl 1):S10 http://www.biomedcentral.com/1471-2164/13/S1/S10 PROCEEDINGS Open Access Identification of gene-oriented exon orthology between human and mouse Gloria C-L Fu1,2,3, Wen-chang Lin3* From The Tenth Asia Pacific Bioinformatics Conference (APBC 2012) Melbourne, Australia. 17-19 January 2012 Abstract Background: Gene orthology has been well studied in the evolutionary area and is thought to be an important implication to functional genome annotations. As the accumulation of transcriptomic data, alternative splicing is taken into account in the assignments of gene orthologs and the orthology is suggested to be further considered at transcript level. Whether gene or transcript orthology, exons are the basic units that represent the whole gene structure; however, there is no any reported study on how to build exon level orthology in a whole genome scale. Therefore, it is essential to establish a gene-oriented exon orthology dataset. Results: Using a customized pipeline, we first build exon orthologous relationships from assigned gene orthologs pairs in two well-annotated genomes: human and mouse. More than 92% of non-overlapping exons have at least one ortholog between human and mouse and only a small portion of them own more than one ortholog. The exons located in the coding region are more conserved in terms of finding their ortholog counterparts. Within the untranslated region, the 5’ UTR seems to have more diversity than the 3’ UTR according to exon orthology designations. Interestingly, most exons located in the coding region are also conserved in length but this conservation phenomenon dramatically drops down in untranslated regions. -

Quantifying the Mechanisms of Domain Gain in Animal Proteins Marija Buljan*, Adam Frankish, Alex Bateman
Buljan et al. Genome Biology 2010, 11:R74 http://genomebiology.com/2010/11/7/R74 RESEARCH Open Access Quantifying the mechanisms of domain gain in animal proteins Marija Buljan*, Adam Frankish, Alex Bateman Abstract Background: Protein domains are protein regions that are shared among different proteins and are frequently functionally and structurally independent from the rest of the protein. Novel domain combinations have a major role in evolutionary innovation. However, the relative contributions of the different molecular mechanisms that underlie domain gains in animals are still unknown. By using animal gene phylogenies we were able to identify a set of high confidence domain gain events and by looking at their coding DNA investigate the causative mechanisms. Results: Here we show that the major mechanism for gains of new domains in metazoan proteins is likely to be gene fusion through joining of exons from adjacent genes, possibly mediated by non-allelic homologous recombination. Retroposition and insertion of exons into ancestral introns through intronic recombination are, in contrast to previous expectations, only minor contributors to domain gains and have accounted for less than 1% and 10% of high confidence domain gain events, respectively. Additionally, exonization of previously non-coding regions appears to be an important mechanism for addition of disordered segments to proteins. We observe that gene duplication has preceded domain gain in at least 80% of the gain events. Conclusions: The interplay of gene duplication and domain gain demonstrates an important mechanism for fast neofunctionalization of genes. Background molecular mechanisms that underlie gains of new Protein domains are fundamental and largely indepen- domains in animals has been more challenging [3]. -

Setd1 Histone 3 Lysine 4 Methyltransferase Complex Components in Epigenetic Regulation
SETD1 HISTONE 3 LYSINE 4 METHYLTRANSFERASE COMPLEX COMPONENTS IN EPIGENETIC REGULATION Patricia A. Pick-Franke Submitted to the faculty of the University Graduate School in partial fulfillment of the requirements for the degree Master of Science in the Department of Biochemistry and Molecular Biology Indiana University December 2010 Accepted by the Faculty of Indiana University, in partial fulfillment of the requirements for the degree of Master of Science. _____________________________________ David Skalnik, Ph.D., Chair _____________________________________ Kristin Chun, Ph.D. Master’s Thesis Committee _____________________________________ Simon Rhodes, Ph.D. ii DEDICATION This thesis is dedicated to my sons, Zachary and Zephaniah who give me great joy, hope and continuous inspiration. I can only hope that I successfully set a good example demonstrating that one can truly accomplish anything, if you never give up and reach for your dreams. iii ACKNOWLEDGEMENTS I would like to thank my committee members Dr. Skalnik, Dr. Chun and Dr. Rhodes for allowing me to complete this dissertation. They have been incredibly generous with their flexibility. I must make a special thank you to Jeanette McClintock, who willingly gave her expertise in statistical analysis with the Cfp1 microarray data along with encouragement, support and guidance to complete this work. I would like to thank Courtney Tate for her ceaseless willingness to share ideas, and her methods and materials, and Erika Dolbrota for her generous instruction as well as the name of a good doctor. I would also like to acknowledge the superb mentorship of Dr. Jeon Heong Lee, PhD and the contagious passion and excitement for the life of science of Dr. -
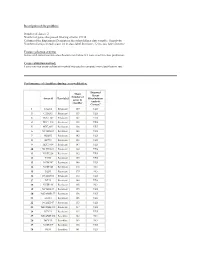
2 Number of Genes That Passed Filtering Criteria
Description of the problem: Number of classes: 2 Number of genes that passed filtering criteria: 25338 Column of the Experiment Descriptors sheet that defines class variable: Sensitivity Number of arrays in each class: 24 in class label Resistant , 32 in class label Sensitive Feature selection criteria: Genes with univariate mis-classfication rate below 0.2 were used for class prediction. Cross-validation method: Leave-one-out cross-validation method was used to compute mis-classification rate. Performance of classifiers during cross-validation. Diagonal Mean Linear Number of Array id Class label Discriminant genes in Analysis classifier Correct? 1 CALU1 Resistant 129 YES 2 CAMA1 Resistant 155 YES 3 HCC1143 Resistant 145 YES 4 HCC1419 Resistant 148 YES 5 HCC2935 Resistant 136 YES 6 NCIH2405 Resistant 148 YES 7 SKBR3 Resistant 142 YES 8 ZR751 Resistant 133 YES 9 HCC1954 Resistant 147 YES 10 NCIH1651 Resistant 139 YES 11 NCIH226 Resistant 142 YES 12 T47D Resistant 139 YES 13 NCIH747 Resistant 146 YES 14 NCIH345 Resistant 216 NO 15 DLD1 Resistant 179 NO 16 NCIH1666 Resistant 154 YES 17 BT20 Resistant 140 YES 18 NCIH196 Resistant 168 NO 19 NCIH1437 Resistant 159 YES 20 MDAMB157 Resistant 158 YES 21 LS123 Resistant 155 YES 22 NCIH2347 Resistant 133 YES 23 MDAMB361 Resistant 137 YES 24 BT474 Resistant 155 YES 25 MDAMB468 Sensitive 182 NO 26 MCF10 Sensitive 169 NO 27 NCIH209 Sensitive 128 YES 28 RKO Sensitive 141 YES 29 HCC70 Sensitive 182 NO 30 LS513 Sensitive 136 YES 31 NCIH1963 Sensitive 122 YES 32 HCC1187 Sensitive 145 YES 33 T84 Sensitive -

Novel Genes Associated with Colorectal Cancer Are Revealed by High Resolution Cytogenetic Analysis in a Patient Specific Manner
Novel Genes Associated with Colorectal Cancer Are Revealed by High Resolution Cytogenetic Analysis in a Patient Specific Manner Hisham Eldai1., Sathish Periyasamy1., Saeed Al Qarni2, Maha Al Rodayyan2, Sabeena Muhammed Mustafa1, Ahmad Deeb3, Ebthehal Al Sheikh2, Mohammed Afzal Khan4, Mishal Johani5, Zeyad Yousef6, Mohammad Azhar Aziz2*. 1 Bioinformatics, King Abdullah International Medical Research Center, Riyadh, Saudi Arabia, 2 Medical Biotechnology, King Abdullah International Medical Research Center, Riyadh, Saudi Arabia, 3 Research Office, King Abdullah International Medical Research Center, Riyadh, Saudi Arabia, 4 Anatomic pathology, National Guard Health Affairs, Riyadh, Saudi Arabia, 5 Endoscopy, National Guard Health Affairs, Riyadh, Saudi Arabia, 6 Surgery, National Guard Health Affairs, Riyadh, Saudi Arabia Abstract Genomic abnormalities leading to colorectal cancer (CRC) include somatic events causing copy number aberrations (CNAs) as well as copy neutral manifestations such as loss of heterozygosity (LOH) and uniparental disomy (UPD). We studied the causal effect of these events by analyzing high resolution cytogenetic microarray data of 15 tumor-normal paired samples. We detected 144 genes affected by CNAs. A subset of 91 genes are known to be CRC related yet high GISTIC scores indicate 24 genes on chromosomes 7, 8, 18 and 20 to be strongly relevant. Combining GISTIC ranking with functional analyses and degree of loss/gain we identify three genes in regions of significant loss (ATP8B1, NARS, and ATP5A1) and eight in regions of gain (CTCFL, SPO11, ZNF217, PLEKHA8, HOXA3, GPNMB, IGF2BP3 and PCAT1) as novel in their association with CRC. Pathway and target prediction analysis of CNA affected genes and microRNAs, respectively indicates TGF-b signaling pathway to be involved in causing CRC. -

Network-Based Analysis of Key Regulatory Genes Implicated in Type
www.nature.com/scientificreports OPEN Network‑based analysis of key regulatory genes implicated in Type 2 Diabetes Mellitus and Recurrent Miscarriages in Turner Syndrome Anam Farooqui1, Alaa Alhazmi2, Shaful Haque3, Naaila Tamkeen4, Mahboubeh Mehmankhah1, Safa Tazyeen1, Sher Ali5 & Romana Ishrat1* The information on the genotype–phenotype relationship in Turner Syndrome (TS) is inadequate because very few specifc candidate genes are linked to its clinical features. We used the microarray data of TS to identify the key regulatory genes implicated with TS through a network approach. The causative factors of two common co‑morbidities, Type 2 Diabetes Mellitus (T2DM) and Recurrent Miscarriages (RM), in the Turner population, are expected to be diferent from that of the general population. Through microarray analysis, we identifed nine signature genes of T2DM and three signature genes of RM in TS. The power‑law distribution analysis showed that the TS network carries scale‑free hierarchical fractal attributes. Through local‑community‑paradigm (LCP) estimation we fnd that a strong LCP is also maintained which means that networks are dynamic and heterogeneous. We identifed nine key regulators which serve as the backbone of the TS network. Furthermore, we recognized eight interologs functional in seven diferent organisms from lower to higher levels. Overall, these results ofer few key regulators and essential genes that we envisage have potential as therapeutic targets for the TS in the future and the animal models studied here may prove useful in the validation of such targets. Te medical systems and scientists throughout the world are under an unprecedented challenge to meet the medical needs of much of the world’s population that are sufering from chromosomal anomalies.