Quantifying the Mechanisms of Domain Gain in Animal Proteins Marija Buljan*, Adam Frankish, Alex Bateman
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gene Expression Profile of THP-1 Cells Treated with Heat-Killed Candida Albicans
Original Article Page 1 of 5 Gene expression profile of THP-1 cells treated with heat-killed Candida albicans Zhi-De Hu1,2*, Ting-Ting Wei1*, Qing-Qin Tang1, Ning Ma1, Li-Li Wang1, Bao-Dong Qin1, Jian-Rong Yin1, Lin Zhou1, Ren-Qian Zhong1 1Department of Laboratory Medicine, Changzheng Hospital, Second Military Medical University, Shanghai 200003, China; 2Department of Laboratory Medicine, General Hospital, Ji’nan Military Region of PLA, Ji’nan 250031, China Contributions: (I) Conception and design: RQ Zhong, L Zhou, ZD Hu; (II) Administrative support: RQ Zhong; (III) Provision of study materials or patients: All authors; (IV) Collection and assembly of data: All authors; (V) Data analysis and interpretation: All authors; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. *These authors contributed equally to this work. Correspondence to: Ren-Qian Zhong, MD, PhD. Department of Laboratory Medicine, Changzheng Hospital, the Second Military Medical University, Shanghai 200003, China. Email: [email protected]; Lin Zhou, MD, PhD. Department of Laboratory Medicine, Changzheng Hospital, the Second Military Medical University, Shanghai 200003, China. Email: [email protected]. Background: Mechanisms under immune response against Candida albicans (C. albicans) remain largely unknown. To better understand the mechanisms of innate immune response against C. albicans, we analyzed the gene expression profile of THP-1 cells stimulated with heat-killed C. albicans. Methods: THP-1 cells were stimulated with heat-killed C. albicans for 9 hours at a ratio of 1:1, and gene expression profile of the cells was analyzed using Whole Human Genome Oligo Microarray. Differentially expressed genes were defined as change folds more than 2 and with statistical significance. -

Nº Ref Uniprot Proteína Péptidos Identificados Por MS/MS 1 P01024
Document downloaded from http://www.elsevier.es, day 26/09/2021. This copy is for personal use. Any transmission of this document by any media or format is strictly prohibited. Nº Ref Uniprot Proteína Péptidos identificados 1 P01024 CO3_HUMAN Complement C3 OS=Homo sapiens GN=C3 PE=1 SV=2 por 162MS/MS 2 P02751 FINC_HUMAN Fibronectin OS=Homo sapiens GN=FN1 PE=1 SV=4 131 3 P01023 A2MG_HUMAN Alpha-2-macroglobulin OS=Homo sapiens GN=A2M PE=1 SV=3 128 4 P0C0L4 CO4A_HUMAN Complement C4-A OS=Homo sapiens GN=C4A PE=1 SV=1 95 5 P04275 VWF_HUMAN von Willebrand factor OS=Homo sapiens GN=VWF PE=1 SV=4 81 6 P02675 FIBB_HUMAN Fibrinogen beta chain OS=Homo sapiens GN=FGB PE=1 SV=2 78 7 P01031 CO5_HUMAN Complement C5 OS=Homo sapiens GN=C5 PE=1 SV=4 66 8 P02768 ALBU_HUMAN Serum albumin OS=Homo sapiens GN=ALB PE=1 SV=2 66 9 P00450 CERU_HUMAN Ceruloplasmin OS=Homo sapiens GN=CP PE=1 SV=1 64 10 P02671 FIBA_HUMAN Fibrinogen alpha chain OS=Homo sapiens GN=FGA PE=1 SV=2 58 11 P08603 CFAH_HUMAN Complement factor H OS=Homo sapiens GN=CFH PE=1 SV=4 56 12 P02787 TRFE_HUMAN Serotransferrin OS=Homo sapiens GN=TF PE=1 SV=3 54 13 P00747 PLMN_HUMAN Plasminogen OS=Homo sapiens GN=PLG PE=1 SV=2 48 14 P02679 FIBG_HUMAN Fibrinogen gamma chain OS=Homo sapiens GN=FGG PE=1 SV=3 47 15 P01871 IGHM_HUMAN Ig mu chain C region OS=Homo sapiens GN=IGHM PE=1 SV=3 41 16 P04003 C4BPA_HUMAN C4b-binding protein alpha chain OS=Homo sapiens GN=C4BPA PE=1 SV=2 37 17 Q9Y6R7 FCGBP_HUMAN IgGFc-binding protein OS=Homo sapiens GN=FCGBP PE=1 SV=3 30 18 O43866 CD5L_HUMAN CD5 antigen-like OS=Homo -

Novel Signature Genes for Human Left Ventricle Cardiomyopathies Identifed by Weighted Co- Expression Network Analysis (WGCNA)
Novel Signature Genes for Human Left Ventricle Cardiomyopathies Identied by Weighted Co- expression Network Analysis (WGCNA) Jiao Tian Kunming Institute of Botany Chinese Academy of Sciences Zhengyuan Wu Kunming Institute of Botany Chinese Academy of Sciences Yaqi Zhang Kunming Institute of Botany Chinese Academy of Sciences Yingying He Yunnan University SHUBAI LIU ( [email protected] ) Kunming Institute of Botany Chinese Academy of Sciences Research Keywords: Heart failure, Cardiomyopathy, Ventricle cardiomyocytes, WGCNA, Signature gene Posted Date: October 8th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-86136/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/29 Abstract Background Cardiomyopathy, a heart disease that arises from different etiologies, brings a huge healthcare burden to the global society. Identication of biomarkers can be very useful for early diagnosis of cardiomyopathy, interruption of the disease procession to heart failure, and decrement of the mortality. Methods Clinical cases of cardiomyopathy were screened out of independently investigations from the genomic database. Exploration of its whole transcription disorder pattern by WGCNA (Weighted Gene Co- expression Network Analysis) to discover the signature genes for different subtypes of cardiomyopathy. The hub genes and key pathways, which are correlated to cardiomyopathy traits, were identied through co-expression and protein-protein interaction (PPI) networks enrichment analysis. Discovered hub genes were blast through the Cardiovascular Disease Portal to verify functions related to human cardiomyopathies. Results Three common axes of signature genes were discovered for ve subtypes of cardiomyopathy: 1) Four genes (MDM4, CFLAR, RPS6KB1, PKD1L2) were common for ischemic and ischemic cardiomyopathy subgroups; 2) Subtypes of cardiomyopathy (ischemic, post. -

Setd1 Histone 3 Lysine 4 Methyltransferase Complex Components in Epigenetic Regulation
SETD1 HISTONE 3 LYSINE 4 METHYLTRANSFERASE COMPLEX COMPONENTS IN EPIGENETIC REGULATION Patricia A. Pick-Franke Submitted to the faculty of the University Graduate School in partial fulfillment of the requirements for the degree Master of Science in the Department of Biochemistry and Molecular Biology Indiana University December 2010 Accepted by the Faculty of Indiana University, in partial fulfillment of the requirements for the degree of Master of Science. _____________________________________ David Skalnik, Ph.D., Chair _____________________________________ Kristin Chun, Ph.D. Master’s Thesis Committee _____________________________________ Simon Rhodes, Ph.D. ii DEDICATION This thesis is dedicated to my sons, Zachary and Zephaniah who give me great joy, hope and continuous inspiration. I can only hope that I successfully set a good example demonstrating that one can truly accomplish anything, if you never give up and reach for your dreams. iii ACKNOWLEDGEMENTS I would like to thank my committee members Dr. Skalnik, Dr. Chun and Dr. Rhodes for allowing me to complete this dissertation. They have been incredibly generous with their flexibility. I must make a special thank you to Jeanette McClintock, who willingly gave her expertise in statistical analysis with the Cfp1 microarray data along with encouragement, support and guidance to complete this work. I would like to thank Courtney Tate for her ceaseless willingness to share ideas, and her methods and materials, and Erika Dolbrota for her generous instruction as well as the name of a good doctor. I would also like to acknowledge the superb mentorship of Dr. Jeon Heong Lee, PhD and the contagious passion and excitement for the life of science of Dr. -
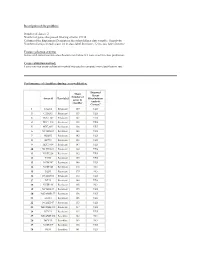
2 Number of Genes That Passed Filtering Criteria
Description of the problem: Number of classes: 2 Number of genes that passed filtering criteria: 25338 Column of the Experiment Descriptors sheet that defines class variable: Sensitivity Number of arrays in each class: 24 in class label Resistant , 32 in class label Sensitive Feature selection criteria: Genes with univariate mis-classfication rate below 0.2 were used for class prediction. Cross-validation method: Leave-one-out cross-validation method was used to compute mis-classification rate. Performance of classifiers during cross-validation. Diagonal Mean Linear Number of Array id Class label Discriminant genes in Analysis classifier Correct? 1 CALU1 Resistant 129 YES 2 CAMA1 Resistant 155 YES 3 HCC1143 Resistant 145 YES 4 HCC1419 Resistant 148 YES 5 HCC2935 Resistant 136 YES 6 NCIH2405 Resistant 148 YES 7 SKBR3 Resistant 142 YES 8 ZR751 Resistant 133 YES 9 HCC1954 Resistant 147 YES 10 NCIH1651 Resistant 139 YES 11 NCIH226 Resistant 142 YES 12 T47D Resistant 139 YES 13 NCIH747 Resistant 146 YES 14 NCIH345 Resistant 216 NO 15 DLD1 Resistant 179 NO 16 NCIH1666 Resistant 154 YES 17 BT20 Resistant 140 YES 18 NCIH196 Resistant 168 NO 19 NCIH1437 Resistant 159 YES 20 MDAMB157 Resistant 158 YES 21 LS123 Resistant 155 YES 22 NCIH2347 Resistant 133 YES 23 MDAMB361 Resistant 137 YES 24 BT474 Resistant 155 YES 25 MDAMB468 Sensitive 182 NO 26 MCF10 Sensitive 169 NO 27 NCIH209 Sensitive 128 YES 28 RKO Sensitive 141 YES 29 HCC70 Sensitive 182 NO 30 LS513 Sensitive 136 YES 31 NCIH1963 Sensitive 122 YES 32 HCC1187 Sensitive 145 YES 33 T84 Sensitive -

Network-Based Analysis of Key Regulatory Genes Implicated in Type
www.nature.com/scientificreports OPEN Network‑based analysis of key regulatory genes implicated in Type 2 Diabetes Mellitus and Recurrent Miscarriages in Turner Syndrome Anam Farooqui1, Alaa Alhazmi2, Shaful Haque3, Naaila Tamkeen4, Mahboubeh Mehmankhah1, Safa Tazyeen1, Sher Ali5 & Romana Ishrat1* The information on the genotype–phenotype relationship in Turner Syndrome (TS) is inadequate because very few specifc candidate genes are linked to its clinical features. We used the microarray data of TS to identify the key regulatory genes implicated with TS through a network approach. The causative factors of two common co‑morbidities, Type 2 Diabetes Mellitus (T2DM) and Recurrent Miscarriages (RM), in the Turner population, are expected to be diferent from that of the general population. Through microarray analysis, we identifed nine signature genes of T2DM and three signature genes of RM in TS. The power‑law distribution analysis showed that the TS network carries scale‑free hierarchical fractal attributes. Through local‑community‑paradigm (LCP) estimation we fnd that a strong LCP is also maintained which means that networks are dynamic and heterogeneous. We identifed nine key regulators which serve as the backbone of the TS network. Furthermore, we recognized eight interologs functional in seven diferent organisms from lower to higher levels. Overall, these results ofer few key regulators and essential genes that we envisage have potential as therapeutic targets for the TS in the future and the animal models studied here may prove useful in the validation of such targets. Te medical systems and scientists throughout the world are under an unprecedented challenge to meet the medical needs of much of the world’s population that are sufering from chromosomal anomalies. -

Wo2018/232195
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2018/232195 Al 20 December 2018 (20.12.2018) W !P O PCT (51) International Patent Classification: MANIAN, Ayshwarya; 77 Massachusetts Ave., Cam A61K 31/00 (2006.01) C07K 16/28 (2006.01) bridge, MA 02139 (US). C07K 14/705 (2006.01) C12Q 1/68 (2018.01) (72) Inventor: ROZENBLATT-ROSEN, Orit; 415 Main C07K 16/24 (2006.01) G06F 19/00 (2018.01) Street, Cambridge, MA 02142 (US). (21) International Application Number: (74) Agent: NIX, F., Brent et al.; Johnson, Marcou & Isaacs, PCT/US20 18/037662 LLC, P.O. Box 691, Hoschton, GA 30548 (US). (22) International Filing Date: (81) Designated States (unless otherwise indicated, for every 14 June 2018 (14.06.2018) kind of national protection available): AE, AG, AL, AM, (25) Filing Language: English AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, (26) Publication Langi English DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, (30) Priority Data: HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, 62/5 19,788 14 June 2017 (14.06.2017) US KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, [US/US]; (71) Applicants: THE BROAD INSTITUTE, INC. OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, 415 Main Street, Cambridge, MA 02142 (US). -

Mouse Uevld Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Uevld Knockout Project (CRISPR/Cas9) Objective: To create a Uevld knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Uevld gene (NCBI Reference Sequence: NM_001040695 ; Ensembl: ENSMUSG00000043262 ) is located on Mouse chromosome 7. 12 exons are identified, with the ATG start codon in exon 1 and the TGA stop codon in exon 12 (Transcript: ENSMUST00000094398). Exon 2~5 will be selected as target site. Cas9 and gRNA will be co-injected into fertilized eggs for KO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Exon 2 starts from about 3.04% of the coding region. Exon 2~5 covers 31.92% of the coding region. The size of effective KO region: ~7790 bp. The KO region does not have any other known gene. Page 1 of 9 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele 5' gRNA region gRNA region 3' 1 2 3 4 5 12 Legends Exon of mouse Uevld Knockout region Page 2 of 9 https://www.alphaknockout.com Overview of the Dot Plot (up) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section upstream of Exon 2 is aligned with itself to determine if there are tandem repeats. Tandem repeats are found in the dot plot matrix. The gRNA site is selected outside of these tandem repeats. Overview of the Dot Plot (down) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section downstream of Exon 5 is aligned with itself to determine if there are tandem repeats. -

Genetic Basis of Lactation and Lactation Efficiency in Pigs Dinesh Moorkattukara Thekkoot Iowa State University
Iowa State University Capstones, Theses and Graduate Theses and Dissertations Dissertations 2014 Genetic basis of lactation and lactation efficiency in pigs Dinesh Moorkattukara Thekkoot Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/etd Part of the Agriculture Commons, and the Animal Sciences Commons Recommended Citation Moorkattukara Thekkoot, Dinesh, "Genetic basis of lactation and lactation efficiency in pigs" (2014). Graduate Theses and Dissertations. 14217. https://lib.dr.iastate.edu/etd/14217 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Genetic basis of lactation and lactation efficiency in pigs by Dinesh Moorkattukara Thekkoot A dissertation submitted to the graduate faculty in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Major: Animal Breeding and Genetics (Quantitative Genetics) Program of Study Committee: Jack C. M. Dekkers, Major Professor Max F. Rothschild Dorian J. Garrick Kenneth J. Stalder Kenneth J. Koehler Iowa State University Ames, Iowa 2014 Copyright © Dinesh Moorkattukara Thekkoot, 2014. All rights reserved. ii TABLE OF CONTENTS Page LIST OF FIGURES iv LIST OF TABLES vii ACKNOWLEDGEMENTS xii ABSTRACT xiii CHAPTER 1. GENERAL INTRODUCTION 1 Introduction 1 Dissertation Organization 4 Review of Literature 5 Conclusion 16 Literature Cited 17 CHAPTER 2. ESTIMATION OF GENETIC PARAMETERS FOR TRAITS ASSOCIATED WITH REPRODUCTION, LACTATION AND EFFICIENCY IN SOWS 22 Abstract 22 Introduction 23 Materials and Methods 24 Results 36 Discussion 50 Conclusion 66 Literature Cited 67 Supplementary Materials 72 CHAPTER 3. -

View U.S. Patent No. 10131950 in PDF
I 1111111111111111 1111111111 1111111111 lllll 111111111111111 111111111111111111 US010131950B2 c12) United States Patent (IO) Patent No.: US 10,131,950 B2 Muir et al. (45) Date of Patent: Nov. 20, 2018 (54) METHOD TO PREDICT HERITABLE DPPA3 Gene Cards, Human Gene Database (Year: 2017). * CANINE NON-CONTACT CRUCIATE Alentorn-Geli et al. (2009). Prevention of non-contact anterioro LIGAMENT RUPTURE curicate ligament injuries in soccer players. Part 1: Mechanisms of injury and underlying risk factors. Knee Surgery, Sports, Traumatol ogy, Arthroscopy. 17:705-729. (71) Applicant: Wisconsin Alumni Research Baird et al.(2014). Genome-wide association study identifies genomic Foundation, Madison, WI (US) regions of association for cruciate ligament rupture in Newfound land dogs. Animal Genetics. 45, 4: 542-549. (72) Inventors: Peter Muir, Madison, WI (US); Beynnon et al. (2006) The relationship between menstrual cycle Lauren Baker, Madison, WI (US) phase and anterior cruciate ligament injury. Am J Sports Med., 34: 757-764. (73) Assignee: Wisconsin Alumni Research Bleedorn et al. (2011). Synovitis in dogs with stable stifle joints and incipient cranial cruciate ligament rupture: A cross-sectional study. Foundation, Madison, WI (US) Veterinary Surgery. 40: 531-543. Chang et al. (2015) Second-generation PLINK: rising to the chal ( *) Notice: Subject to any disclaimer, the term ofthis lenge of larger and richer datasets. GigaScience. 4:7. patent is extended or adjusted under 35 Chen et al. (2011) A genetic risk score combining ten psoriasis risk U.S.C. 154(b) by 80 days. loci improves disease prediction. PLoS One. 6: el9454. Chuang et al. (2014), Radiographic risk factors for contralateral (21) Appl. -

Somatic Genomic Changes in the Formation of Differentiated Thyroid Carcinoma
10.2478/AMB-2020-0036 SOMATIC GENOMIC CHANGES IN THE FORMATION OF DIFFERENTIATED THYROID CARCINOMA K. Vidinov1, R. Dodova2, I. Dimitrova1, A. Mitkova2, A. Shinkov1, R. Kaneva2, R. Kovacheva1 1Department of Endocrinology, Medical Faculty, Medical University – Sofia, Bulgaria 2Molecular Medicine Center, Department of Medical Chemistry and Biochemistry, Medical Faculty, Medical University – Sofia, Bulgaria Abstract. Globally, the diffuse goiter affects more than 10% of the population and in some regions is endemic. Thyroid nodules are found in approximately 5% of the population us- ing the oldest method for thyroid examination – palpation. When performing ultrasound screening, this percentage increases significantly and reaches between 20 and 75% of the total population. Thyroid carcinoma is a rare malignancy and accounts for up to 1% of all malignant tumors. It is the most common endocrine cancer and is clinically manifested as a thyroid nodule. Somatic mutations play an important role in its development. Differentia- tion of benign and malignant thyroid nodules is of great importance due to the different therapeutic approach. Therefore, new diagnostic tools are sought to help distinguish the two. Despite the progress in our knowledge of carcinogenesis in recent years, a number of key issues still remain unanswered. The establishment of new rare somatic mutations can improve pre-surgical diagnosis and optimize post-operative strategies for the treatment of thyroid carcinoma. Next-generation sequencing (NGS) allows for extensive mutation and genome rearrangements tracking. The results obtained with NGS provide the basis for the development of new approach for systematic genetic screening, at prevention, early diag- nosis, accurate prognosis, and targeted therapy of this disorder. -

Targeted Next Generation Sequencing Identifies Somatic Mutations and Gene Fusions in Papillary Thyroid Carcinoma
www.impactjournals.com/oncotarget/ Oncotarget, 2017, Vol. 8, (No. 28), pp: 45784-45792 Research Paper Targeted next generation sequencing identifies somatic mutations and gene fusions in papillary thyroid carcinoma Zheming Lu1,*, Yujie Zhang2,*, Dongdong Feng2, Jindong Sheng2, Wenjun Yang1,3 and Baoguo Liu2 1Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education), Laboratory of Biochemistry and Molecular Biology, Beijing, China 2Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education), Department of Head and Neck Surgery, Peking University Cancer Hospital & Institute, Beijing, China 3Key Laboratory of Fertility Preservation and Maintenance (Ministry of Education), Cancer Institute of the General Hospital, Ningxia Medical University, Yinchuan, China *These authors have contributed equally to this work Correspondence to: Wenjun Yang, email: [email protected] Baoguo Liu, email: [email protected] Keywords: papillary thyroid carcinoma (PTC), cancer panel, fusion gene, somatic mutation Received: February 08, 2017 Accepted: April 04, 2017 Published: April 25, 2017 Copyright: Lu et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License 3.0 (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT 138 papillary thyroid carcinoma (PTC) samples were assessed for somatic mutation profile and fusion genes by targeted resequencing using a cancer panel (ThyGenCapTM) targeting 244 cancer-related genes and 20 potential fusion genes. At least one genetic alteration (including mutations and fusion genes) was observed in 118/138 (85.5%) samples. The most frequently mutated gene was BRAF V600E (57.2%). Moreover, we identified 11 fusion genes including eight previously reported ones and three novel fusion genes, UEVLD-RET, OSBPL9-BRAF, and SQSTM1-NTRK3.