UC San Francisco Electronic Theses and Dissertations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genome-Wide Association Study of Increasing Suicidal Ideation During Antidepressant Treatment in the GENDEP Project
The Pharmacogenomics Journal (2012) 12, 68–77 & 2012 Macmillan Publishers Limited. All rights reserved 1470-269X/12 www.nature.com/tpj ORIGINAL ARTICLE Genome-wide association study of increasing suicidal ideation during antidepressant treatment in the GENDEP project NPerroud1,2,3,RUher1, Suicidal thoughts during antidepressant treatment have been the focus of 1 2,3,4 several candidate gene association studies. The aim of the present genome- MYM Ng , M Guipponi , wide association study was to identify additional genetic variants involved in 5 6 JHauser, N Henigsberg , increasing suicidal ideation during escitalopram and nortriptyline treatment. WMaier7,OMors8, A total of 706 adult participants of European ancestry, treated for major M Gennarelli9, M Rietschel10, depression with escitalopram or nortriptyline over 12 weeks in the Genome- DSouery11, MZ Dernovsek12, Based Therapeutic Drugs for Depression (GENDEP) study were genotyped 13 14 with Illumina Human 610-Quad Beadchips (Illumina, San Diego, CA, USA). AS Stamp ,MLathrop , A total of 244 subjects experienced an increase in suicidal ideation AFarmer1, G Breen1,KJAitchison1, during follow-up. The genetic marker most significantly associated with CM Lewis1,IWCraig1 increasing suicidality (8.28 Â 10À7) was a single-nucleotide polymorphism and P McGuffin1 (SNP; rs11143230) located 30 kb downstream of a gene encoding guanine deaminase (GDA) on chromosome 9q21.13. Two suggestive drug-specific 1MRC Social, Genetic and Developmental Psychiatry associations within KCNIP4 (Kv channel-interacting protein 4; chromosome Centre, Institute of Psychiatry, King’s College 4p15.31) and near ELP3 (elongation protein 3 homolog; chromosome London, London, UK; 2Department of Psychiatry, 8p21.1) were found in subjects treated with escitalopram. -

Analysis of Trans Esnps Infers Regulatory Network Architecture
Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2014 © 2014 Anat Kreimer All rights reserved ABSTRACT Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer eSNPs are genetic variants associated with transcript expression levels. The characteristics of such variants highlight their importance and present a unique opportunity for studying gene regulation. eSNPs affect most genes and their cell type specificity can shed light on different processes that are activated in each cell. They can identify functional variants by connecting SNPs that are implicated in disease to a molecular mechanism. Examining eSNPs that are associated with distal genes can provide insights regarding the inference of regulatory networks but also presents challenges due to the high statistical burden of multiple testing. Such association studies allow: simultaneous investigation of many gene expression phenotypes without assuming any prior knowledge and identification of unknown regulators of gene expression while uncovering directionality. This thesis will focus on such distal eSNPs to map regulatory interactions between different loci and expose the architecture of the regulatory network defined by such interactions. We develop novel computational approaches and apply them to genetics-genomics data in human. We go beyond pairwise interactions to define network motifs, including regulatory modules and bi-fan structures, showing them to be prevalent in real data and exposing distinct attributes of such arrangements. We project eSNP associations onto a protein-protein interaction network to expose topological properties of eSNPs and their targets and highlight different modes of distal regulation. -

A Context-Dependent Role for the RNF146 Ubiquitin Ligase in Wingless/Wnt Signaling in Drosophila
| INVESTIGATION A Context-Dependent Role for the RNF146 Ubiquitin Ligase in Wingless/Wnt Signaling in Drosophila Zhenghan Wang,* Ofelia Tacchelly-Benites,* Geoffrey P. Noble,* Megan K. Johnson,* Jean-Philippe Gagné,† Guy G. Poirier,† and Yashi Ahmed*,1 *Department of Molecular and Systems Biology, Norris Cotton Cancer Center, Geisel School of Medicine at Dartmouth College HB7400, Hanover, New Hampshire 03755 and †Centre de Recherche du Centre Hospitalier Universitaire de Québec-Université Laval, CHUL Pavilion, Axe Oncologie, Québec G1V 4G2, Canada ABSTRACT Aberrant activation of the Wnt signal transduction pathway triggers the development of colorectal cancer. The ADP-ribose polymerase Tankyrase (TNKS) mediates proteolysis of Axin—a negative regulator of Wnt signaling—and provides a promising ther- apeutic target for Wnt-driven diseases. Proteolysis of TNKS substrates is mediated through their ubiquitination by the poly-ADP-ribose (pADPr)-dependent RING-domain E3 ubiquitin ligase RNF146/Iduna. Like TNKS, RNF146 promotes Axin proteolysis and Wnt pathway activation in some cultured cell lines, but in contrast with TNKS, RNF146 is dispensable for Axin degradation in colorectal carcinoma cells. Thus, the contexts in which RNF146 is essential for TNKS-mediated Axin destabilization and Wnt signaling remain uncertain. Herein, we tested the requirement for RNF146 in TNKS-mediated Axin proteolysis and Wnt pathway activation in a range of in vivo settings. Using null mutants in Drosophila, we provide genetic and biochemical evidence that Rnf146 and Tnks function in the same proteolysis pathway in vivo. Furthermore, like Tnks, Drosophila Rnf146 promotes Wingless signaling in multiple developmental contexts by buffering Axin levels to ensure they remain below the threshold at which Wingless signaling is inhibited. -

UNIVERSITY of CALIFORNIA, SAN DIEGO Functional Analysis of Sall4
UNIVERSITY OF CALIFORNIA, SAN DIEGO Functional analysis of Sall4 in modulating embryonic stem cell fate A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Molecular Pathology by Pei Jen A. Lee Committee in charge: Professor Steven Briggs, Chair Professor Geoff Rosenfeld, Co-Chair Professor Alexander Hoffmann Professor Randall Johnson Professor Mark Mercola 2009 Copyright Pei Jen A. Lee, 2009 All rights reserved. The dissertation of Pei Jen A. Lee is approved, and it is acceptable in quality and form for publication on microfilm and electronically: ______________________________________________________________ ______________________________________________________________ ______________________________________________________________ ______________________________________________________________ Co-Chair ______________________________________________________________ Chair University of California, San Diego 2009 iii Dedicated to my parents, my brother ,and my husband for their love and support iv Table of Contents Signature Page……………………………………………………………………….…iii Dedication…...…………………………………………………………………………..iv Table of Contents……………………………………………………………………….v List of Figures…………………………………………………………………………...vi List of Tables………………………………………………….………………………...ix Curriculum vitae…………………………………………………………………………x Acknowledgement………………………………………………….……….……..…...xi Abstract………………………………………………………………..…………….....xiii Chapter 1 Introduction ..…………………………………………………………………………….1 Chapter 2 Materials and Methods……………………………………………………………..…12 -

Tumor-Associated Antigens Identified Early in Mouse Mammary Tumor Development Can Be Effective Vaccine Targets
Vaccine 37 (2019) 3552–3561 Contents lists available at ScienceDirect Vaccine journal homepage: www.elsevier.com/locate/vaccine Tumor-associated antigens identified early in mouse mammary tumor development can be effective vaccine targets ⇑ Sasha E. Stanton a, , Ekram Gad a, Lauren R. Corulli a, Hailing Lu a,1, Mary L. Disis a a Cancer Vaccine Institute, University of Washington, Seattle WA, 98109, USA article info abstract Article history: Breast cancer vaccines composed of antigens identified by serological analysis of cDNA expression Received 25 June 2018 libraries (SEREX) induce antigen specific immune responses in patients but have had disappointing clin- Received in revised form 5 April 2019 ical benefits. While many attempts to modify the adjuvants and vaccine method have been tried, one Accepted 9 May 2019 issue not addressed was whether the SEREX tumor-associated antigens identified from late stages of dis- Available online 21 May 2019 ease were ideal targets. We questioned in the transgenic TgMMTV-neu mouse model whether the antigen repertoire is distinct between early and late stage breast cancer and whether the antigens identified via Keywords: SEREX from transgenic mice with early or late stage tumors would elicit differential anti-tumor effects to Breast cancer prevention address this question. Vaccine antigens Th1 Three early stage antigens, Pdhx, Stk39, and Otud6B, were identified from a SEREX screen of mice prior DNA vaccines to development of palpable lesions. Formulated into a vaccine, each early antigen inhibited tumor growth Mouse mammary tumor models (p < 0.0001). The antigens identified from mice with late stage tumors (Swap70, Gsn, and Arhgef2) were unable to inhibit tumor growth when used as vaccines (for example Gsn p = 0.26). -

Contiguous Deletion of the NDP, MAOA, MAOB, and EFHC2 Genes in a Patient with Norrie Disease, Severe Psychomotor Retardation and Myoclonic Epilepsy
ß 2007 Wiley-Liss, Inc. American Journal of Medical Genetics Part A 143A:916–920 (2007) Contiguous Deletion of the NDP, MAOA, MAOB, and EFHC2 Genes in a Patient With Norrie Disease, Severe Psychomotor Retardation and Myoclonic Epilepsy L. Rodriguez-Revenga,1,2 I. Madrigal,1,2 L.S. Alkhalidi,3 L. Armengol,4 E. Gonza´lez,4 C. Badenas,1,2 X. Estivill,1,5 and M. Mila`1,2* 1Biochemistry and Molecular Genetics Department, Hospital Clı´nic, Barcelona, Spain 2IDIBAPS (Institut d’Investigacions Biome`diques August Pi i Sunyer), Barcelona, Spain 3Department of Health and Medical Services, Rashid Hospital, Dubai, United Arab Emirates 4Genes and Disease Programme, Centre for Genomic Regulation (CRG), Barcelona Biomedical Research Park, Barcelona, Spain 5Department of Experimental and Health Sciences, Universitat Pompeu Fabra (UPF), Barcelona, Spain Received 20 February 2006; Accepted 7 September 2006 Norrie disease (ND) is an X-linked disorder, inherited as a Clinical features of the proband include bilateral retinal recessive trait that, therefore, mostly affects males. The gene detachment, microcephaly, severe psychomotor retardation responsible for ND, called NDP, maps to the short arm of without verbal language skills acquired, and epilepsy. The chromosome X (Xp11.4-p11.3). We report here an atypical identification and molecular characterization of this case case of ND, consisting of a patient harboring a large reinforces the idea of a new contiguous gene syndrome that submicroscopic deletion affecting not only the NDP gene would explain the complex phenotype shared by atypical but also the MAOA, MAOB, and EFHC2 genes. Microarray ND patients. ß 2007 Wiley-Liss, Inc. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

BTG2: a Rising Star of Tumor Suppressors (Review)
INTERNATIONAL JOURNAL OF ONCOLOGY 46: 459-464, 2015 BTG2: A rising star of tumor suppressors (Review) BIjING MAO1, ZHIMIN ZHANG1,2 and GE WANG1 1Cancer Center, Institute of Surgical Research, Daping Hospital, Third Military Medical University, Chongqing 400042; 2Department of Oncology, Wuhan General Hospital of Guangzhou Command, People's Liberation Army, Wuhan, Hubei 430070, P.R. China Received September 22, 2014; Accepted November 3, 2014 DOI: 10.3892/ijo.2014.2765 Abstract. B-cell translocation gene 2 (BTG2), the first 1. Discovery of BTG2 in TOB/BTG gene family gene identified in the BTG/TOB gene family, is involved in many biological activities in cancer cells acting as a tumor The TOB/BTG genes belong to the anti-proliferative gene suppressor. The BTG2 expression is downregulated in many family that includes six different genes in vertebrates: TOB1, human cancers. It is an instantaneous early response gene and TOB2, BTG1 BTG2/TIS21/PC3, BTG3 and BTG4 (Fig. 1). plays important roles in cell differentiation, proliferation, DNA The conserved domain of BTG N-terminal contains two damage repair, and apoptosis in cancer cells. Moreover, BTG2 regions, named box A and box B, which show a high level of is regulated by many factors involving different signal path- homology to the other domains (1-5). Box A has a major effect ways. However, the regulatory mechanism of BTG2 is largely on cell proliferation, while box B plays a role in combination unknown. Recently, the relationship between microRNAs and with many target molecules. Compared with other family BTG2 has attracted much attention. MicroRNA-21 (miR-21) members, BTG1 and BTG2 have an additional region named has been found to regulate BTG2 gene during carcinogenesis. -

Supplemental Table 1. Complete Gene Lists and GO Terms from Figure 3C
Supplemental Table 1. Complete gene lists and GO terms from Figure 3C. Path 1 Genes: RP11-34P13.15, RP4-758J18.10, VWA1, CHD5, AZIN2, FOXO6, RP11-403I13.8, ARHGAP30, RGS4, LRRN2, RASSF5, SERTAD4, GJC2, RHOU, REEP1, FOXI3, SH3RF3, COL4A4, ZDHHC23, FGFR3, PPP2R2C, CTD-2031P19.4, RNF182, GRM4, PRR15, DGKI, CHMP4C, CALB1, SPAG1, KLF4, ENG, RET, GDF10, ADAMTS14, SPOCK2, MBL1P, ADAM8, LRP4-AS1, CARNS1, DGAT2, CRYAB, AP000783.1, OPCML, PLEKHG6, GDF3, EMP1, RASSF9, FAM101A, STON2, GREM1, ACTC1, CORO2B, FURIN, WFIKKN1, BAIAP3, TMC5, HS3ST4, ZFHX3, NLRP1, RASD1, CACNG4, EMILIN2, L3MBTL4, KLHL14, HMSD, RP11-849I19.1, SALL3, GADD45B, KANK3, CTC- 526N19.1, ZNF888, MMP9, BMP7, PIK3IP1, MCHR1, SYTL5, CAMK2N1, PINK1, ID3, PTPRU, MANEAL, MCOLN3, LRRC8C, NTNG1, KCNC4, RP11, 430C7.5, C1orf95, ID2-AS1, ID2, GDF7, KCNG3, RGPD8, PSD4, CCDC74B, BMPR2, KAT2B, LINC00693, ZNF654, FILIP1L, SH3TC1, CPEB2, NPFFR2, TRPC3, RP11-752L20.3, FAM198B, TLL1, CDH9, PDZD2, CHSY3, GALNT10, FOXQ1, ATXN1, ID4, COL11A2, CNR1, GTF2IP4, FZD1, PAX5, RP11-35N6.1, UNC5B, NKX1-2, FAM196A, EBF3, PRRG4, LRP4, SYT7, PLBD1, GRASP, ALX1, HIP1R, LPAR6, SLITRK6, C16orf89, RP11-491F9.1, MMP2, B3GNT9, NXPH3, TNRC6C-AS1, LDLRAD4, NOL4, SMAD7, HCN2, PDE4A, KANK2, SAMD1, EXOC3L2, IL11, EMILIN3, KCNB1, DOK5, EEF1A2, A4GALT, ADGRG2, ELF4, ABCD1 Term Count % PValue Genes regulation of pathway-restricted GDF3, SMAD7, GDF7, BMPR2, GDF10, GREM1, BMP7, LDLRAD4, SMAD protein phosphorylation 9 6.34 1.31E-08 ENG pathway-restricted SMAD protein GDF3, SMAD7, GDF7, BMPR2, GDF10, GREM1, BMP7, LDLRAD4, phosphorylation -

RING-Type E3 Ligases: Master Manipulators of E2 Ubiquitin-Conjugating Enzymes and Ubiquitination☆
Biochimica et Biophysica Acta 1843 (2014) 47–60 Contents lists available at ScienceDirect Biochimica et Biophysica Acta journal homepage: www.elsevier.com/locate/bbamcr Review RING-type E3 ligases: Master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination☆ Meredith B. Metzger a,1, Jonathan N. Pruneda b,1, Rachel E. Klevit b,⁎, Allan M. Weissman a,⁎⁎ a Laboratory of Protein Dynamics and Signaling, Center for Cancer Research, National Cancer Institute, 1050 Boyles Street, Frederick, MD 21702, USA b Department of Biochemistry, Box 357350, University of Washington, Seattle, WA 98195, USA article info abstract Article history: RING finger domain and RING finger-like ubiquitin ligases (E3s), such as U-box proteins, constitute the vast Received 5 March 2013 majority of known E3s. RING-type E3s function together with ubiquitin-conjugating enzymes (E2s) to medi- Received in revised form 23 May 2013 ate ubiquitination and are implicated in numerous cellular processes. In part because of their importance in Accepted 29 May 2013 human physiology and disease, these proteins and their cellular functions represent an intense area of study. Available online 6 June 2013 Here we review recent advances in RING-type E3 recognition of substrates, their cellular regulation, and their varied architecture. Additionally, recent structural insights into RING-type E3 function, with a focus on im- Keywords: RING finger portant interactions with E2s and ubiquitin, are reviewed. This article is part of a Special Issue entitled: U-box Ubiquitin–Proteasome System. Guest Editors: Thomas Sommer and Dieter H. Wolf. Ubiquitin ligase (E3) Published by Elsevier B.V. Ubiquitin-conjugating enzyme (E2) Protein degradation Catalysis 1. -

Noelia Díaz Blanco
Effects of environmental factors on the gonadal transcriptome of European sea bass (Dicentrarchus labrax), juvenile growth and sex ratios Noelia Díaz Blanco Ph.D. thesis 2014 Submitted in partial fulfillment of the requirements for the Ph.D. degree from the Universitat Pompeu Fabra (UPF). This work has been carried out at the Group of Biology of Reproduction (GBR), at the Department of Renewable Marine Resources of the Institute of Marine Sciences (ICM-CSIC). Thesis supervisor: Dr. Francesc Piferrer Professor d’Investigació Institut de Ciències del Mar (ICM-CSIC) i ii A mis padres A Xavi iii iv Acknowledgements This thesis has been made possible by the support of many people who in one way or another, many times unknowingly, gave me the strength to overcome this "long and winding road". First of all, I would like to thank my supervisor, Dr. Francesc Piferrer, for his patience, guidance and wise advice throughout all this Ph.D. experience. But above all, for the trust he placed on me almost seven years ago when he offered me the opportunity to be part of his team. Thanks also for teaching me how to question always everything, for sharing with me your enthusiasm for science and for giving me the opportunity of learning from you by participating in many projects, collaborations and scientific meetings. I am also thankful to my colleagues (former and present Group of Biology of Reproduction members) for your support and encouragement throughout this journey. To the “exGBRs”, thanks for helping me with my first steps into this world. Working as an undergrad with you Dr. -
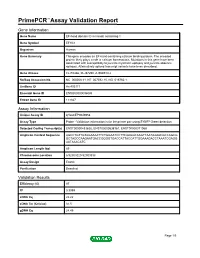
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name EF-hand domain (C-terminal) containing 1 Gene Symbol EFHC1 Organism Human Gene Summary This gene encodes an EF-hand-containing calcium binding protein. The encoded protein likely plays a role in calcium homeostasis. Mutations in this gene have been associated with susceptibility to juvenile myoclonic epilepsy and juvenile absence epilepsy. Alternatively spliced transcript variants have been described. Gene Aliases FLJ10466, FLJ37290, dJ304B14.2 RefSeq Accession No. NC_000006.11, NT_007592.15, NG_016760.1 UniGene ID Hs.403171 Ensembl Gene ID ENSG00000096093 Entrez Gene ID 114327 Assay Information Unique Assay ID qHsaCEP0049958 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000433625, ENST00000538167, ENST00000371068 Amplicon Context Sequence AGCCTGTTGTAGAAAATTCTGGAATCCTTCAAGGCAAGTTAATAAAACGCCAGCG GCTAGCCAAGAATGACCGGGGTGACCATTACCATTGGAAAGACCTAAATCGAGG AATAAACATC Amplicon Length (bp) 89 Chromosome Location 6:52303220-52303338 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 97 R2 0.9999 cDNA Cq 22.22 cDNA Tm (Celsius) 81.5 gDNA Cq 24.45 Page 1/5 PrimePCR™Assay Validation Report Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report EFHC1, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification