Development of Genomic Resources for Ornamental Lilies (Lilium L.)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Liliaceae S.L. (Lily Family)
Liliaceae s.l. (Lily family) Photo: Ben Legler Photo: Hannah Marx Photo: Hannah Marx Lilium columbianum Xerophyllum tenax Trillium ovatum Liliaceae s.l. (Lily family) Photo: Yaowu Yuan Fritillaria lanceolata Ref.1 Textbook DVD KRR&DLN Erythronium americanum Allium vineale Liliaceae s.l. (Lily family) Herbs; Ref.2 Stems often modified as underground rhizomes, corms, or bulbs; Flowers actinomorphic; 3 sepals and 3 petals or 6 tepals, 6 stamens, 3 carpels, ovary superior (or inferior). Tulipa gesneriana Liliaceae s.l. (Lily family) “Liliaceae” s.l. (sensu lato: “in the broad sense”) - Lily family; 288 genera/4950 species, including Lilium, Allium, Trillium, Tulipa; This family is treated in a very broad sense in this class, as in the Flora of the Pacific Northwest. The “Liliaceae” s.l. taught in this class is not monophyletic. It is apparent now that the family should be treated in a narrower sense and some of the members should form their own families. Judd et al. recognize 15+ families: Agavaceae, Alliaceae, Amarylidaceae, Asparagaceae, Asphodelaceae, Colchicaceae, Dracaenaceae (Nolinaceae), Hyacinthaceae, Liliaceae, Melanthiaceae, Ruscaceae, Smilacaceae, Themidaceae, Trilliaceae, Uvulariaceae and more!!! (see web reading “Consider the Lilies”) Iridaceae (Iris family) Photo: Hannah Marx Photo: Hannah Marx Iris pseudacorus Iridaceae (Iris family) Photo: Yaowu Yuan Photo: Yaowu Yuan Sisyrinchium douglasii Sisyrinchium sp. Iridaceae (Iris family) Iridaceae - 78 genera/1750 species, Including Iris, Gladiolus, Sisyrinchium. Herbs, aquatic or terrestrial; Underground stems as rhizomes, bulbs, or corms; Leaves alternate, 2-ranked and equitant Ref.3 (oriented edgewise to the stem; Gladiolus italicus Flowers actinomorphic or zygomorphic; 3 sepals and 3 petals or 6 tepals; Stamens 3; Ovary of 3 fused carpels, inferior. -

Guide to the Flora of the Carolinas, Virginia, and Georgia, Working Draft of 17 March 2004 -- LILIACEAE
Guide to the Flora of the Carolinas, Virginia, and Georgia, Working Draft of 17 March 2004 -- LILIACEAE LILIACEAE de Jussieu 1789 (Lily Family) (also see AGAVACEAE, ALLIACEAE, ALSTROEMERIACEAE, AMARYLLIDACEAE, ASPARAGACEAE, COLCHICACEAE, HEMEROCALLIDACEAE, HOSTACEAE, HYACINTHACEAE, HYPOXIDACEAE, MELANTHIACEAE, NARTHECIACEAE, RUSCACEAE, SMILACACEAE, THEMIDACEAE, TOFIELDIACEAE) As here interpreted narrowly, the Liliaceae constitutes about 11 genera and 550 species, of the Northern Hemisphere. There has been much recent investigation and re-interpretation of evidence regarding the upper-level taxonomy of the Liliales, with strong suggestions that the broad Liliaceae recognized by Cronquist (1981) is artificial and polyphyletic. Cronquist (1993) himself concurs, at least to a degree: "we still await a comprehensive reorganization of the lilies into several families more comparable to other recognized families of angiosperms." Dahlgren & Clifford (1982) and Dahlgren, Clifford, & Yeo (1985) synthesized an early phase in the modern revolution of monocot taxonomy. Since then, additional research, especially molecular (Duvall et al. 1993, Chase et al. 1993, Bogler & Simpson 1995, and many others), has strongly validated the general lines (and many details) of Dahlgren's arrangement. The most recent synthesis (Kubitzki 1998a) is followed as the basis for familial and generic taxonomy of the lilies and their relatives (see summary below). References: Angiosperm Phylogeny Group (1998, 2003); Tamura in Kubitzki (1998a). Our “liliaceous” genera (members of orders placed in the Lilianae) are therefore divided as shown below, largely following Kubitzki (1998a) and some more recent molecular analyses. ALISMATALES TOFIELDIACEAE: Pleea, Tofieldia. LILIALES ALSTROEMERIACEAE: Alstroemeria COLCHICACEAE: Colchicum, Uvularia. LILIACEAE: Clintonia, Erythronium, Lilium, Medeola, Prosartes, Streptopus, Tricyrtis, Tulipa. MELANTHIACEAE: Amianthium, Anticlea, Chamaelirium, Helonias, Melanthium, Schoenocaulon, Stenanthium, Veratrum, Toxicoscordion, Trillium, Xerophyllum, Zigadenus. -

Genome Annotation of Fritillaria Agrestis Bac Clone
GENOME ANNOTATION OF FRITILLARIA AGRESTIS BAC CLONE ___________________ A University Thesis Presented to the Faculty of California State University, East Bay ___________________ In Partial Fulfillment of the Requirements for the Degree Master of Science in Biological Science ___________________ By Rajhalutshimi Narayanaswamy September 2015 GENOME ANNOTATION OF FRITILLARJA AGRESTIS BAC CLONE By Rajhalutshimi Narayanaswamy Date: A~~u, ~o1s ii Acknowledgments I would like to thank my advisors Dr.Chris Baysdorfer, Dr.Claudia Udhe-Stone and Dr.Kenneth Curr for their support, guidance and encouragement that helped me to complete this project successfully. I would also like to thank my family members who have been supportive during the course of my project. iii Table of Contents Acknowledgments.............................................................................................................. iii List of Figures ................................................................................................................... vii 1 Introduction ...............................................................................................................1 1.1 Genome size variation ...........................................................................................1 1.2 Brief outline of the mechanisms of genome size variation ...................................2 1.3 Repetitious DNA ...................................................................................................3 1.4 Annotation ...........................................................................................................21 -

1 the Global Flower Bulb Industry
1 The Global Flower Bulb Industry: Production, Utilization, Research Maarten Benschop Hobaho Testcentrum Hillegom, The Netherlands Rina Kamenetsky Department of Ornamental Horticulture Agricultural Research Organization The Volcani Center Bet Dagan 50250, Israel Marcel Le Nard Institut National de la Recherche Agronomique 29260 Ploudaniel, France Hiroshi Okubo Laboratory of Horticultural Science Kyushu University 6-10-1 Hakozaki, Higashi-ku Fukuoka 812-8581, Japan August De Hertogh Department of Horticultural Science North Carolina State University Raleigh, NC 29565-7609, USA COPYRIGHTED MATERIAL I. INTRODUCTION II. HISTORICAL PERSPECTIVES III. GLOBALIZATION OF THE WORLD FLOWER BULB INDUSTRY A. Utilization and Development of Expanded Markets Horticultural Reviews, Volume 36 Edited by Jules Janick Copyright Ó 2010 Wiley-Blackwell. 1 2 M. BENSCHOP, R. KAMENETSKY, M. LE NARD, H. OKUBO, AND A. DE HERTOGH B. Introduction of New Crops C. International Conventions IV. MAJOR AREAS OF RESEARCH A. Plant Breeding and Genetics 1. Breeders’ Right and Variety Registration 2. Hortus Bulborum: A Germplasm Repository 3. Gladiolus 4. Hyacinthus 5. Iris (Bulbous) 6. Lilium 7. Narcissus 8. Tulipa 9. Other Genera B. Physiology 1. Bulb Production 2. Bulb Forcing and the Flowering Process 3. Morpho- and Physiological Aspects of Florogenesis 4. Molecular Aspects of Florogenesis C. Pests, Physiological Disorders, and Plant Growth Regulators 1. General Aspects for Best Management Practices 2. Diseases of Ornamental Geophytes 3. Insects of Ornamental Geophytes 4. Physiological Disorders of Ornamental Geophytes 5. Exogenous Plant Growth Regulators (PGR) D. Other Research Areas 1. Specialized Facilities and Equipment for Flower Bulbs52 2. Transportation of Flower Bulbs 3. Forcing and Greenhouse Technology V. MAJOR FLOWER BULB ORGANIZATIONS A. -

Elucidating and Mining the Tulipa and Lilium Transcriptomes
Plant Mol Biol DOI 10.1007/s11103-016-0508-1 Elucidating and mining the Tulipa and Lilium transcriptomes Natalia M. Moreno-Pachon1 · Hendrika A. C. F. Leeggangers1 · Harm Nijveen1,2 · Edouard Severing2,3 · Henk Hilhorst4 · Richard G. H. Immink1 Received: 26 April 2016 / Accepted: 27 June 2016 © The Author(s) 2016. This article is published with open access at Springerlink.com Abstract Genome sequencing remains a challenge for resource via a user-friendly web-based interface. The species with large and complex genomes containing exten- Illumina HiSeq 2000 platform was applied and the tran- sive repetitive sequences, of which the bulbous and mono- scribed RNA was sequenced from a collection of different cotyledonous plants tulip and lily are examples. In such lily and tulip tissues, respectively. In order to obtain good a case, sequencing of only the active part of the genome, transcriptome coverage and to facilitate effective data min- represented by the transcriptome, is a good alternative to ing, assembly was done using different filtering parameters obtain information about gene content. In this study we for clearing out contamination and noise of the RNAseq aimed to generate a high quality transcriptome of tulip datasets. This analysis revealed limitations of commonly and lily and to make this data available as an open-access applied methods and parameter settings used in de novo transcriptome assembly. The final created transcriptomes are publicly available via a user friendly Transcriptome browser (http://www.bioinformatics.nl/bulbs/db/species/ Natalia M. Moreno-Pachon, Hendrika A.C.F. Leeggangers and Harm index). The usefulness of this resource has been exempli- Nijveen have equally contributed equally to this work. -

Historical Survey of Easter Lily Name in Association with Lilium Longiflorum
® Floriculture and Ornamental Biotechnology ©2012 Global Science Books Historical Survey of Easter Lily Name in Association with Lilium longiflorum Eisuke Matsuo Professor Emeritus of Kyushu University, 1198-68, Tsuko, Ogori-shi 838-0102, Japan Correspondence : [email protected] ABSTRACT Based on the English dictionaries from around 1900, the author traces back how Lilium longiflorum came to be referred to as the Easter lily. The term “Easter lily” was given to any lily-like flowering species that bloomed around the Easter day, being called Easter flowers. Among them, L. candidum became the most famous one known as “Easter lily”. The evidence suggests that when more easily forcible bulb production began in the Bermuda Islands, the superiority of L. longiflorum over L. candidum in forcing ability and price, might have led to an explosive promotion of the usage of L. longiflorum as the Easter flower instead of L. candidum. Thus, the name "Easter lily" came to be transferred from L. candidum to L. longiflorum. _____________________________________________________________________________________________________________ Keywords: Easter decoration, Easter flower, Lilium candidum, Madonna lily CONTENTS INTRODUCTION.......................................................................................................................................................................................... 9 EASTER FLOWER AND EASTER LILY................................................................................................................................................... -

Overexpression of Lilium Formosanum MADS-Box (LFMADS) Causing
International Journal of Molecular Sciences Article Overexpression of Lilium formosanum MADS-box (LFMADS) Causing Floral Defects While Promoting Flowering in Arabidopsis thaliana, Whereas Only Affecting Floral Transition Time in Nicotiana tabacum Wan-Yu Liao 1,†, Lee-Fong Lin 2,† ID , Ming-Der Lin 3, Sheng-Che Hsieh 2, Althea Yi-Shan Li 2, Yueh-Shiah Tsay 4 and Ming-Lun Chou 1,2,* ID 1 Institute of Medical Sciences, Tzu-Chi University, Hualien 97004, Taiwan; [email protected] 2 Department of Life Sciences, Tzu-Chi University, Hualien 97004, Taiwan; [email protected] (L.-F.L.); [email protected] (S.-C.H.); [email protected] (A.Y.-S.L.) 3 Department of Molecular Biology and Human Genetics, Tzu-Chi University, Hualien 97004, Taiwan; [email protected] 4 Division of Crop Improvement, Hualien District Agricultural Research and Extension Station, Council of Agriculture, Executive Yuan, Hualien 97365, Taiwan; [email protected] * Correspondence: [email protected]; Tel.: +886-3-8465615; Fax: +886-3-8572526 † These authors contributed equally to this study. Received: 30 June 2018; Accepted: 26 July 2018; Published: 29 July 2018 Abstract: The Formosa lily (Lilium formosanum) is one of the most common horticultural species in Taiwan. To explore gene regulation involved in this species, we used transcriptome analysis to generate PH-FB (mixed floral buds) and PH-LF (mature leaves) datasets. Combination of the PH-FB and PH-LF constructed a de novo assembly of the ALL dataset, including 18,041 contigs and 23,807 unigenes by Nr, GO, COG, and KEGG databases. -

Botanical Priority Guidebook
Botanical Priority Protection Areas Alameda and Contra Costa Counties the East Bay Regional Park District. However, certain BPPAs include Hills have been from residential development. public parcels or properties with other conservation status. These are cases where land has been conserved since the creation of these boundaries or where potential management decisions have the poten- Following this initial mapping effort, the East Bay Chap- \ ntroduction tial to negatively affect an area’s botanical resources. Additionally, ter’s Conservation Committee began to utilize the con- each acre within these BPPAs represents a potential area of high pri- cept in draft form in key local planning efforts. Lech ority. Both urban and natural settings are included within these Naumovich, the chapter’s Conservation Analyst staff The lands that comprise the East Bay Chapter are located at the convergence boundaries, therefore, they are intended to be considered as areas person, showcased the map set in forums such as the of the San Francisco Bay, the North and South Coast Ranges, the Sacra- warranting further scrutiny due to the abundance of nearby sensitive BAOSC’s Upland Habitat Goals Project and the Green mento-San Joaquin Delta, and the San Joaquin Valley. The East Bay Chapter botanical resources supported by high quality habitat within each E A S T B A Y Vision Group (in association with Greenbelt Alliance); area supports a unique congregation of ecological conditions and native BPPA. Although a parcel, available for preservation through fee title C N P S East Bay Regional Park District’s Master Plan Process; plants. Based on historic botanical collections, the pressures from growth- purchase or conservation easement, may be located within the and local municipalities. -

TG/115/4 ORIGINAL: English DATE: 2006-04-05 INTERNATIONAL UNION for the PROTECTION of NEW VARIETIES of PLANTS GENEVA
E TG/115/4 ORIGINAL: English DATE: 2006-04-05 INTERNATIONAL UNION FOR THE PROTECTION OF NEW VARIETIES OF PLANTS GENEVA * TULIP UPOV Code: TULIP Tulipa L. GUIDELINES FOR THE CONDUCT OF TESTS FOR DISTINCTNESS, UNIFORMITY AND STABILITY * Alternative Names: Botanical name English French German Spanish Tulipa L. Tulip Tulipe Tulpe Tulipán The purpose of these guidelines (“Test Guidelines”) is to elaborate the principles contained in the General Introduction (document TG/1/3), and its associated TGP documents, into detailed practical guidance for the harmonized examination of distinctness, uniformity and stability (DUS) and, in particular, to identify appropriate characteristics for the examination of DUS and production of harmonized variety descriptions. ASSOCIATED DOCUMENTS These Test Guidelines should be read in conjunction with the General Introduction and its associated TGP documents. * These names were correct at the time of the introduction of these Test Guidelines but may be revised or updated. [Readers are advised to consult the UPOV Code, which can be found on the UPOV Website (www.upov.int), for the latest information.] TG/115/4 Tulip, 2006-04-05 - 2 - TABLE OF CONTENTS PAGE 1. SUBJECT OF THESE TEST GUIDELINES ....................................................................................................3 2. MATERIAL REQUIRED..................................................................................................................................3 3. METHOD OF EXAMINATION........................................................................................................................3 -
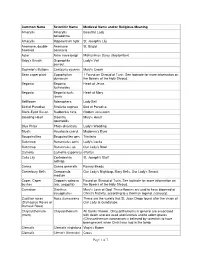
Of 7 Common Name Scientific Name Medieval Name And/Or Religious Meaning Amaryllis Amaryllis Belladonna Beautiful Lady
Common Name Scientific Name Medieval Name and/or Religious Meaning Amaryllis Amaryllis Beautiful Lady belladonna Amaryllis Hippeastrum hybr. St. Joseph's Lily Anemone, double- Anemone St. Brigid flowered coronaria Aster Aster nova-belgii Michaelmas Daisy (September) Baby's Breath Gypsophila Lady's Veil panicul. Bachelor's Buttons Centauria cyannis Mary's Crown Bean caper plant Zygophyllum ? Found on Shroud of Turin. See footnote for more information on dumosum the flowers of the Holy Shroud. Begonia Begonia Heart of Jesus fuchsioides Begonia Begonia fuch. Heart of Mary rosea Bellflower Adenophera Lady Bell Bird of Paradise Streliztia reginae Bird of Paradise Black-Eyed Susan Rudbeckia hirta Golden Jerusalem Bleeding Heart Dicentra Mary's Heart spectabilis Blue Phlox Phlox divaricata Lady's Wedding Bluets Houstonia caerul. Madonna's Eyes Bougainvillea Bougainvillea gen. Trinitaria Buttercup Ranunculus acris Lady's Locks Buttercup Ranunculus sp. Our Lady's Bowl Camelia Camellia (japonica) (Purity) Calla Lily Zantedeshia St. Joseph's Staff aethiop. Canna Canna generalis Rosary Beads Canterbury Bells Campanula Our Lady's Nightcap, Mary Bells, Our Lady's Smock medium Caper, Caper Capparis spinosa Found on Shroud of Turin. See footnote for more information on bushes (var. aegyptia) the flowers of the Holy Shroud. Carnation Dianthus Mary's Love of God. These flowers are said to have bloomed at caryophyllus Christ's Nativity, according to a German legend. (January) Castilian roses Rosa damascena These are the variety that St. Juan Diego found after the vision of (Damascus Roses or Our Lady at Guadalupe. Damask Rose) Chrysanthemum Chrysanthemum All Saints' Flower. Chrysanthemums in general are associated (mum) with death and are used and funerals and to adorn graves (Chrysanthemum coronarium is believed by scientists to have been present when Christ was laid in the tomb. -

LILIUM) PRODUCTION Faculty of Science, Department of Biology, University of Oulu
BIOTECHNOLOGICAL APPROACHES VELI-PEKKA PELKONEN IN LILY (LILIUM) PRODUCTION Faculty of Science, Department of Biology, University of Oulu OULU 2005 VELI-PEKKA PELKONEN BIOTECHNOLOGICAL APPROACHES IN LILY (LILIUM) PRODUCTION Academic Dissertation to be presented with the assent of the Faculty of Science, University of Oulu, for public discussion in Kuusamonsali (Auditorium YB210), Linnanmaa, on April 15th, 2005, at 12 noon OULUN YLIOPISTO, OULU 2005 Copyright © 2005 University of Oulu, 2005 Supervised by Professor Anja Hohtola Professor Hely Häggman Reviewed by Professor Anna Bach Professor Risto Tahvonen ISBN 951-42-7658-2 (nid.) ISBN 951-42-7659-0 (PDF) http://herkules.oulu.fi/isbn9514276590/ ISSN 0355-3191 http://herkules.oulu.fi/issn03553191/ OULU UNIVERSITY PRESS OULU 2005 Pelkonen, Veli-Pekka, Biotechnological approaches in lily (Lilium) production Faculty of Science, Department of Biology, University of Oulu, P.O.Box 3000, FIN-90014 University of Oulu, Finland 2005 Oulu, Finland Abstract Biotechnology has become a necessity, not only in research, but also in the culture and breeding of lilies. Various methods in tissue culture and molecular breeding have been applied to the production of commercially important lily species and cultivars. However, scientific research data of such species and varieties that have potential in the northern climate is scarce. In this work, different biotechnological methods were developed and used in the production and culture of a diversity of lily species belonging to different taxonomic groups. The aim was to test and develop further the existing methods in plant biotechnology for the developmental work and the production of novel hardy lily cultivars for northern climates. -

Easter Lily Growth and Development
Easter Lily Growth and Development 1985 Lot aClef - uNIPR TqTE .` cam V /rr sl 81. ,! l9' Technical Bulletin 148 AGRICULTURAL EXPERIMENT STATION Oregon State University Corvallis, Oregon Easter Lily Growth and Development ACKNOWLEDGMENTS We wish to acknowledge the help and encouragement of colleagues concernedwith lily bulb growing, handling, and forcing at Oregon State over the years. Faculty members including L. T.Blaney,O. C.Compton. Ralph Garren, L. H.Fuchigami, J. C.Green, P. J. Breen, and J. R. Stang have made important contributions to our knowledge of Lilium physiology. Graduate students in the Department of Horticulture have contributed much of thedetail to thisknowledge. These graduates include Vernon Clarkson (M.S. 1951),P. C. Lin(M.S. 1968). Dave Hartley (Ph.D.1968). S. Y. Wang(Ph.D.1969).Raj Bahadur(Ph.D. 1969), W. R. McCorkle(B.S. 1970, Missouri),K. D. Gray(M.S. 1974), B. J. Tomasovic(B.S.1974),and Y. T. Wang(M.S. 1981). We are especially grateful tothe FredC. Cloeckncr Foundation for its many years of financial support of the Easter lily research project and in preparation of this publication. The same is true for the Pacific Bulb Growers' Association, which has provided a field research station, a technician for its maintenance, graduate student support, and operating expenses at the station for more than 25 years. We would also mention the valuable contributions made by the Agricultural Extension Services of both Oregon and California through their able representatives Walt Schroeder (Curry County, Oregon) and John Lenz (Del Norte County, California). We are indebted to H.