04Chapter3.Pdf 577Kb
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Routine Multipesticide Analysis Orbitrap
Routine Multi-Pesticide Residue Analysis by Orbitrap MS Technology Osama Abu-Nimreh CMD Sales Support Specialist MECEC , Dubai The world leader in serving science Challenges of Pesticide-Residues Analysis • Sample variability (matrix) • Different compound characteristics • Large number of samples • Hundreds of analytes monitored • Low levels controlled • Baby food (MRL for all pesticides = 0.01 mg/kg) • Fast response required 2 Former Pesticide Multi-Residue Method Setup . Extraction Acetonitrile, Ethyl Mostly replaced acetate, Methanol... by QuEChERS today . Clean-up GPC, SPE, LLE, LC . Determination GC, LC, GC-MS, LC-MS, Thermo Scientific™ QuEChERS™ GC-MS/MS, LC-MS/MS... method 3 Simplified Extraction Procedure Applied 10 g of sample is weighed into Quechers extraction tube + 20 mL of water + 10 mL of ACN shaking 10 min Centrifugation 5 min @ 5000 rpm Injection to LC-HRAM 4 Consumables Used Consumables/Chemicals Part Number Acetonitrile A/0638/17 QuEChERS extraction tube, 50 mL, 250 pack 60105-216 QuEChERS pouches, 50 pack 60105-344 Apparatus/Columns Part Number Horizontal shaker 1069-3391 Horizontal shaker plate 1053-0102 Thermo Scientific™ Barnstead™ EASYpure™II water 3125753 Thermo Scientific™ Heraeus™ Fresco™ 17 micro centrifuge 3208590 Thermo Scientific™ Accucore™ aQ column 100x2.1, 2.6 µm 17326-102130 5 Improving QuEChERS Extraction Tips & Tricks: • Dry food (cereals/dried food, < 25 % water content): • Addition of water to enable adequate partitioning and reducing interaction of pesticides with matrix • Food containing fat/wax (avocado/oil): • After extraction step add a freezing out step and transfer supernatant to clean-up tube • More clean-up might be needed of raw extract (PSA+C18) • Food containing complex matrix (tea/spices) • Additional clean-up with GCB might be necessary (potential loss of planar structure pesticides like thiabendazole) • Acidic food (citrus): • Adjust pH (5-5.5) to increase recovery (e.g. -

Application of Dried Blood Spots for the Determination of Organophosphorus Pesticides by GC-MS/MS
UNIVERSIDADE DA BEIRA INTERIOR Ciências Application of dried blood spots for the determination of organophosphorus pesticides by GC-MS/MS Sofia Pires Seixo Soares Dissertação para obtenção do Grau de Mestre em Bioquímica (2º ciclo de estudos) Orientador: Doutor Mário Jorge Dinis Barroso Co-orientador: Professora Doutora Maria Eugenia Gallardo Alba Covilhã, junho de 2018 Agradecimentos A realização de uma dissertação de mestrado é o culminar de muitas horas de estudo, reflexão e trabalho pessoal, mas representa também uma etapa que não seria possível sem o contributo fundamental de várias pessoas. À Doutora Eugénia Gallardo e ao Doutor Mário Barroso, como excelentes mentores que são, quero agradecer toda a orientação, motivação, incentivo e confiança. Toda a ajuda e partilha de experiência e conhecimento durante todo este percurso. Ao Tiago Rosado, pela imprescindível ajuda e disponibilidade constante, bem como pela amizade. À Joana Gonçalves e ao Ângelo Luís um agradecimento especial pela amizade e carinho demonstrado ao longo deste ano. A todos, quero agradecer pelo bom ambiente proporcionado. Aos meus pais Ana Soares e José Soares, a quem dedico este feito, por me darem a possibilidade de concretizar conquistas como esta, pelo apoio incondicional, por sempre terem acreditado em mim e por fazerem parte deste momento. Aos meus avós e restante família, por todo o apoio e preocupação. Um agradecimento particular à Inês Ramos, à Rita Marques, à Maria Cunha e ao Pedro Batista pelo companheirismo, amizade e apoio de há tantos anos. E por último, mas nunca menos importante, um enorme obrigada ao meu namorado António Fernandes por toda a paciência, pelo incansável apoio, carinho e ajuda. -

Prohibited and Restricted Pesticides List Fair Trade USA® Agricultural Production Standard Version 1.1.0
Version 1.1.0 Prohibited and Restricted Pesticides List Fair Trade USA® Agricultural Production Standard Version 1.1.0 Introduction Through the implementation of our standards, Fair Trade USA aims to promote sustainable livelihoods and safe working conditions, protection of the environment, and strong, transparent supply chains.. Our standards work to limit negative impacts on communities and the environment. All pesticides can be potentially hazardous to human health and the environment, both on the farm and in the community. They can negatively affect the long-term sustainability of agricultural livelihoods. The Fair Trade USA Agricultural Production Standard (APS) seeks to minimize these risks from pesticides by restricting the use of highly hazardous pesticides and enhancing the implementation of risk mitigation practices for lower risk pesticides. This approach allows greater flexibility for producers, while balancing controls on impacts to human and environmental health. This document lists the pesticides that are prohibited or restricted in the production of Fair Trade CertifiedTM products, as required in Objective 4.4.2 of the APS. It also includes additional rules for the use of restricted pesticides. Purpose The purpose of this document is to outline the rules which prohibit or restrict the use of hazardous pesticides in the production of Fair Trade Certified agricultural products. Scope • The Prohibited and Restricted Pesticides List (PRPL) applies to all crops certified against the Fair Trade USA Agricultural Production Standard (APS). • Restrictions outlined in this list apply to active ingredients in any pesticide used by parties included in the scope of the Certificate while handling Fair Trade Certified products. -

Recommended Classification of Pesticides by Hazard and Guidelines to Classification 2019 Theinternational Programme on Chemical Safety (IPCS) Was Established in 1980
The WHO Recommended Classi cation of Pesticides by Hazard and Guidelines to Classi cation 2019 cation Hazard of Pesticides by and Guidelines to Classi The WHO Recommended Classi The WHO Recommended Classi cation of Pesticides by Hazard and Guidelines to Classi cation 2019 The WHO Recommended Classification of Pesticides by Hazard and Guidelines to Classification 2019 TheInternational Programme on Chemical Safety (IPCS) was established in 1980. The overall objectives of the IPCS are to establish the scientific basis for assessment of the risk to human health and the environment from exposure to chemicals, through international peer review processes, as a prerequisite for the promotion of chemical safety, and to provide technical assistance in strengthening national capacities for the sound management of chemicals. This publication was developed in the IOMC context. The contents do not necessarily reflect the views or stated policies of individual IOMC Participating Organizations. The Inter-Organization Programme for the Sound Management of Chemicals (IOMC) was established in 1995 following recommendations made by the 1992 UN Conference on Environment and Development to strengthen cooperation and increase international coordination in the field of chemical safety. The Participating Organizations are: FAO, ILO, UNDP, UNEP, UNIDO, UNITAR, WHO, World Bank and OECD. The purpose of the IOMC is to promote coordination of the policies and activities pursued by the Participating Organizations, jointly or separately, to achieve the sound management of chemicals in relation to human health and the environment. WHO recommended classification of pesticides by hazard and guidelines to classification, 2019 edition ISBN 978-92-4-000566-2 (electronic version) ISBN 978-92-4-000567-9 (print version) ISSN 1684-1042 © World Health Organization 2020 Some rights reserved. -
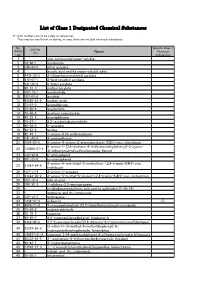
List of Class 1 Designated Chemical Substances
List of Class 1 Designated Chemical Substances *1:CAS numbers are to be solely as references. They may be insufficient or lacking, in case there are multiple chemical substances. No. Specific Class 1 CAS No. (PRTR Chemical (*1) Name Law) Substances 1 - zinc compounds(water-soluble) 2 79-06-1 acrylamide 3 140-88-5 ethyl acrylate 4 - acrylic acid and its water-soluble salts 5 2439-35-2 2-(dimethylamino)ethyl acrylate 6 818-61-1 2-hydroxyethyl acrylate 7 141-32-2 n-butyl acrylate 8 96-33-3 methyl acrylate 9 107-13-1 acrylonitrile 10 107-02-8 acrolein 11 26628-22-8 sodium azide 12 75-07-0 acetaldehyde 13 75-05-8 acetonitrile 14 75-86-5 acetone cyanohydrin 15 83-32-9 acenaphthene 16 78-67-1 2,2'-azobisisobutyronitrile 17 90-04-0 o-anisidine 18 62-53-3 aniline 19 82-45-1 1-amino-9,10-anthraquinone 20 141-43-5 2-aminoethanol 21 1698-60-8 5-amino-4-chloro-2-phenylpyridazin-3(2H)-one; chloridazon 5-amino-1-[2,6-dichloro-4-(trifluoromethyl)phenyl]-3-cyano- 22 120068-37-3 4[(trifluoromethyl)sulfinyl]pyrazole; fipronil 23 123-30-8 p-aminophenol 24 591-27-5 m-aminophenol 4-amino-6-tert-butyl-3-methylthio-1,2,4-triazin-5(4H)-one; 25 21087-64-9 metribuzin 26 107-11-9 3-amino-1-propene 27 41394-05-2 4-amino-3-methyl-6-phenyl-1,2,4-triazin-5(4H)-one; metamitron 28 107-18-6 allyl alcohol 29 106-92-3 1-allyloxy-2,3-epoxypropane 30 - n-alkylbenzenesulfonic acid and its salts(alkyl C=10-14) 31 - antimony and its compounds 32 120-12-7 anthracene 33 1332-21-4 asbestos ○ 34 4098-71-9 3-isocyanatomethyl-3,5,5-trimethylcyclohexyl isocyanate 35 78-84-2 isobutyraldehyde -

United States Patent (19) 11 Patent Number: 5,703,064 Yokoi Et Al
US005703064A United States Patent (19) 11 Patent Number: 5,703,064 Yokoi et al. 45) Date of Patent: Dec. 30, 1997 54 PESTICIDAL COMBINATIONS FOREIGN PATENT DOCUMENTS 75 Inventors: Shinji Yokoi; Akira Nishida, both of 0 196524 10/1986 European Pat. Of.. Shiga-ken; Tokio Obata; Kouichi Golka, both of Ube, all of Japan OTHER PUBLICATIONS 73) Assignees: Sankyo Company, Limited, Tokyo; Worthing et al, The Pesticide Manual, 9th Ed. (1991), pp. Ube industries Ltd., Ube, both of 747 and 748. Japan L.C. Gaughan et al., "Pesticide interactions: effects of orga nophosphorus pesticides on the metabolism, toxicity, and 21 Appl. No.: 405,795 persistence of selected pyrethroid insecticides". Chemical Abstracts, vol. 94, No. 9, 1981, No. 59740k of Pestic. 22 Filed: Mar 16, 1995 Biochem. Physio., vol. 14, No. 1, 1980, pp. 81-85. 30 Foreign Application Priority Data I. Ishaaya et al., "Cypermethrin synergism by pyrethroid esterase inhibitors in adults of the whitefly Bemisia tabaci". Mar 16, 1994 JP Japan ............................ HE6045.405 Chemical Abstracts, vol. 107, No. 9, 1987, No. 72818y of (51) Int. Cl................. A01N 43/54; A01N 57/00 Pestic Biochem. Physiol., vol. 28, No. 2, 1987, pp. 155-162. 52 U.S. C. ......................................... 51480; 514/256 (58) Field of Search ..................................... 514/80, 256 Primary Examiner Allen J. Robinson Attorney, Agent, or Firm-Frishauf, Holtz, Goodman, 56 References Cited Langer & Chick, P.C. U.S. PATENT DOCUMENTS 57 ABSTRACT 4,374,833 2/1983 Badmin et al. ...................... 424/225 Combinations of the known compound pyrimidifen with 4,845,097 7/1989 Matsumoto et al... 514/234.2 phosphorus-containing pesticides have a synergistic pesti 4,935,516 6/1990 Ataka et al. -

NMP-Free Formulations of Neonicotinoids
(19) & (11) EP 2 266 400 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 29.12.2010 Bulletin 2010/52 A01N 43/40 (2006.01) A01N 43/86 (2006.01) A01N 47/40 (2006.01) A01N 51/00 (2006.01) (2006.01) (2006.01) (21) Application number: 09305544.0 A01P 7/00 A01N 25/02 (22) Date of filing: 15.06.2009 (84) Designated Contracting States: (72) Inventors: AT BE BG CH CY CZ DE DK EE ES FI FR GB GR • Gasse, Jean-Jacques HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL 27600 Saint-Aubin-Sur-Gaillon (FR) PT RO SE SI SK TR • Duchamp, Guillaume Designated Extension States: 92230 Gennevilliers (FR) AL BA RS • Cantero, Maria 92230 Gennevilliers (FR) (71) Applicant: NUFARM 92233 Gennevelliers (FR) (74) Representative: Cabinet Plasseraud 52, rue de la Victoire 75440 Paris Cedex 09 (FR) (54) NMP-free formulations of neonicotinoids (57) The invention relates to NMP-free liquid formulation comprising at least one nicotinoid and at least one aprotic polar component selected from the group comprising the compounds of formula I, II or III below, and mixtures thereof, wherein R1 and R2 independently represent H or an alkyl group having less than 5 carbons, preferably a methyl group, and n represents an integer ranging from 0 to 5, and to their applications. EP 2 266 400 A1 Printed by Jouve, 75001 PARIS (FR) EP 2 266 400 A1 Description Technical Field of the invention 5 [0001] The invention relates to novel liquid formulations of neonicotinoids and to their use for treating plants, for protecting plants from pests and/or for controlling pests infestation. -

(Annex 2) Pesticide for Analysis
(Annex 2) Pesticide for analysis No. Pesticide Compound No. Pesticide Compound 644 XMC XMC 430 Nitrothal-isopropyl Nitrothal-isopropyl 43 Azaconazole Azaconazole 433 Norflurazon Norflurazon 14 Acetochlor Acetochlor 68 Bioallethrin Bioallethrin 42 Atrazine Atrazine 479 Picolinafen Picolinafen 31 Ametryn Ametryn 482 Piperophos Piperophos 27 Allethrin Allethrin 516 Pyrazophos Pyrazophos 351 Isazophos Isazophos 520 Pyridafenthion Pyridafenthion 359 Isoxathion Isoxathion 642 Vinclozolin Vinclozolin 355 Isoprothiolane Isoprothiolane 252 Fenamiphos Fenamiphos 348 Iprobenfos(IBP) Iprobenfos(IBP) 260 Fenothiocarb Fenothiocarb 335 Imazamethabenz methyl ester Imazametha benzmethyl ester 470 Phenothrin Phenothrin 237 Ethion Ethion 254 Fenbuconazole Fenbuconazole 238 Ethofumesate Ethofumesate 264 Fenpropimorph Fenpropimorph α-Endosulfan 310 Fthalide Fthalide 226 α-Endosulfan β-Endosulfan 80 Bupirimate Bupirimate 445 Oxadiazon Oxadiazon 81 Buprofezin Buprofezin 446 Oxadixyl Oxadixyl 273 Flamplop-methyl Flamplop-methyl 456 Oxyfluorfen Oxyfluorfen 276 Fluacrypyrim Fluacrypyrim 96 Carfentrazone-ethyl Carfentrazone-ethyl 301 Flutriafol Flutriafol 91 Carbofuran Carbofuran 289 Flumioxazin Flumioxazin 530 Quinoxyfen Quinoxyfen 288 Flumiclorac pentyl Flumiclorac pentyl 529 Quinoclamine Quinoclamine 500 Propachlor Propachlor 531 Quintozene Quintozene 501 Propanil Propanil 135 Clomazone Clomazone 503 Propargite Propargite 126 Chlorthal-dimethyl Chlorthal-dimethyl 508 Propyzamide Propyzamide 124 Chlorpyrifos-methyl Chlorpyrifos-methyl 484 Profenofos Profenofos -
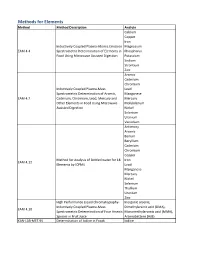
Method Description
Methods for Elements Method Method Description Analyte Calcium Copper Iron Inductively Coupled Plasma-Atomic Emission Magnesium EAM 4.4 Spectrometric Determination of Elements in Phosphorus Food Using Microwave Assisted Digestion Potassium Sodium Strontium Zinc Arsenic Cadmium Chromium Inductively Coupled Plasma-Mass Lead Spectrometric Determination of Arsenic, Manganese EAM 4.7 Cadmium, Chromium, Lead, Mercury and Mercury Other Elements in Food Using Microwave Molybdenum Assisted Digestion Nickel Selenium Uranium Vanadium Antimony Arsenic Barium Beryllium Cadmium Chromium Copper Method for Analysis of Bottled water for 18 Iron EAM 4.12 Elements by ICPMS Lead Manganese Mercury Nickel Selenium Thallium Uranium Zinc High Performance Liquid Chromatography- Inorganic arsenic, Inductively Coupled Plasma-Mass Dimethylarsinic acid (DMA), EAM 4.10 Spectrometric Determination of Four Arsenic Monomethylarsonic acid (MMA), Species in Fruit Juice Arsenobetaine (AsB) KAN-LAB-MET.95 Determination of Iodine in Foods Iodine Methods for Radionuclides Method Method Description Analyte Determination of Strontium-90 in Foods by WEAC.RN.METHOD.2.0 Strontium-90 Internal Gas-Flow Proportional Counting Americium-241 Cesium-134 Cesium-137 Determination of Gamma-Ray Emitting Cobalt-60 WEAC.RN.METHOD.3.0 Radionuclides in Foods by High-Purity Potassium-40 Germanium Spectrometry Radium-226 Ruthenium-103 Ruthenium-106 Thorium-232 Methods for Pesticides/Industrial Chemicals Method Method Description Analyte Extraction Method: Analysis of Pesticides KAN-LAB-PES.53 and -

Analysis of Golf Course Pesticides Using Triple Quadrupole LC/MS/MS
LAAN-A-LM-E051 Liquid Chromatography Mass Spectrometry No.C80 Analysis of Golf Course Pesticides Using Triple Quadrupole LC/MS/MS (LCMS-8030) Many kinds of pesticides are used to maintain golf notification issued on September 29, 2010 increased course turf, and because golf courses are often the list to 72 pesticides and made it a requirement to located in mountain corridors and near water sources, conduct simultaneous analysis of the chemicals using pollution of nearby fresh water sources and tap water higher sensitivity instruments. from golf course waste water is of concern. With this Of those 72 pesticides, we conducted simultaneous increased concern over the environment, in 1990 analysis of 21 of the remaining 44 substances Japan's Ministry of the Environment issued the specified to be evaluated by LC/MS using the LCMS- "Provisional Guidance Indicators for the Prevention of 8030, and in addition, we conducted monitor testing of Water Pollution from Pesticides Used in Golf Courses" golf course wastewater. The results are presented (hereafter, Guidance Indicators), in which indicator here. values were specified for 21 pesticides. Then, a The MRM chromatograms of 21 golf course pesticides the quantitation results. Six consecutive are shown in Fig. 1, and calibration curves for typical measurements were conducted for each compound at golf course pesticides are shown in Fig. 2. Excellent a concentration of 0.5 μg/L, and the area repeatability linearity was obtained for all substances within the %RSD was less than 10 % in all cases. The quantitation concentration range of 0.5 to 100 μg/L. Table 1 shows limit (calculated with S/N = 10) ranged from 0.06 to the parameters used in the MRM mode, in addition to 0.50 μg/L. -

Metabolism of Isoxathion, O,O-Diethyl O-(5-Phenyl-3-Isoxazolyl)
Agr. Biol. Chem., 39 (11), 21372143, 1975 Metabolism of Isoxathion, O,O-Diethyl O-(5-Phenyl-3-isoxazolyl)- phosphorothionate in Plants Mitsuru ANDO,Yukiko IWASAKIand Masayuki NAKAGAWA SankyoCo., Ltd., AgriculturalChemicals Research Laboratories 2-58, l-chome, Hiromachi, Shinagawa-ku, Tokyo, Japan Received May 15, 1975 The absorption and metabolism of the insecticide, Isoxathion, on bean, cabbage and chinese cabbage plants were examined using carbon-14 labeled compound. Isoxathion penetrated into plant tissues was hydrolyzed to produce 3-hydroxy-5-phenylisoxazole, which was then rapidly converted to water soluble compounds. Among them, 3-(ƒÀ-3-D-glucopyranosyl oxy)-5-phenylisoxazole, 2-(ƒÀ-D-glucopyranosyl)-5-phenyl-4-isoxazolin-3-one and 2-(ƒÀ-D-gluco pyranosyl)-5-p-hydroxy-phenyl-4-isoxazolin-3-one were unequivocally identified as the major metabolites. Another metabolic pathway of 3-hydroxy-5-phenylisoxazole via a reductive cleavage of the isoxazole ring to form benzoic acid was negligible. Isoxathion, Karphos_??_, O, O-diethyl O-(5- phenylisoxazole (I), 2-(2•L,3•L,4•L,6•L-tetra-O-acetyl-ƒÀ-D- glucopyranosyl)-5-phenyl-4-isoxazolin-3-one (II), 3- phenyl-3-isoxazolyl)-phosphorothionate, is a (ƒÀ-D-glucopyranosyloxy)-5-phenylisoxazole (III) and 2- registered insecticide of a broad spectrum.'' ƒÀ-D-glucopyranosyl)-5-phenyl-4-isoxazolin-3-one (IV) The toxicity of the compound is as follows: were synthesized as follows: a mixture of 60g (ca. acute (oral) LD,0 to rats is 112 mg/kg" and 0.15M) of 2,3,4,6-tetra-O-acetyl-ƒ¿-D-glucopyranosyl non-effect level to rats in 90 days consecutive bromide," 24.2g (0.15M) of 3-hydroxy-5-phenyliso xazole, 16g (ca. -

Research and Special Programs Admin., DOT § 172.101
Research and Special Programs Admin., DOT § 172.101 TABLE 2 TO APPENDIX AÐRADIONUCLIDESÐ the class of the material must be determined Continued in accordance with § 173.2a of this sub- chapter. 3. This appendix contains two columns. (2)Ð (3)ÐReportable Atomic The first column, entitled ``S.M.P.'' (for se- (1)ÐRadionuclide Num- Quantity (RQ) ber Ci (TBq) vere marine pollutants), identifies whether a material is a severe marine pollutant. If the Ytterbium-169 .......................... 70 10 (.37) letters ``PP'' appear in this column for a ma- Ytterbium-175 .......................... 70 100 (3.7) terial, the material is a severe marine pol- Ytterbium-177 .......................... 70 1000 (37) lutant, otherwise it is not. The second col- Ytterbium-178 .......................... 70 1000 (37) umn, entitled ``Marine Pollutant'' , lists the Yttrium-86 ................................ 39 10 (.37) Yttrium-86m ............................. 39 1000 (37) marine pollutants. Yttrium-87 ................................ 39 10 (.37) 4. If a material not listed in this appendix Yttrium-88 ................................ 39 10 (.37) meets the criteria for a marine pollutant, as Yttrium-90 ................................ 39 10 (.37) provided in the General Introduction of the Yttrium-90m ............................. 39 100 (3.7) IMDG Code, Guidelines for the Identification Yttrium-91 ................................ 39 10 (.37) of Harmful Substances in Packaged Form, Yttrium-91m ............................. 39 1000 (37) Yttrium-92 ................................ 39 100 (3.7) the material may be transported as a marine Yttrium-93 ................................ 39 100 (3.7) pollutant in accordance with the applicable Yttrium-94 ................................ 39 1000 (37) requirements of this subchapter. Yttrium-95 ................................ 39 1000 (37) 5. If approved by the Associate Adminis- Zinc-62 ..................................... 30 100 (3.7) trator for Hazardous Materials Safety, a ma- Zinc-63 ....................................