Synthesis and Characterization of Amide and Urea Receptor Systems
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Paclitaxel-Loaded Glycyrrhizic Acid Micelles
Molecules 2015, 20, 4337-4356; doi:10.3390/molecules20034337 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Article Bioavailability Enhancement of Paclitaxel via a Novel Oral Drug Delivery System: Paclitaxel-Loaded Glycyrrhizic Acid Micelles Fu-Heng Yang †, Qing Zhang †, Qian-Ying Liang, Sheng-Qi Wang, Bo-Xin Zhao, Ya-Tian Wang, Yun Cai and Guo-Feng Li * Department of Pharmacy, Nanfang Hospital, Southern Medical University, Guangzhou 510515, China; E-Mails: [email protected] (F.-H.Y.); [email protected] (Q.Z.); [email protected] (Q.-Y.L.); [email protected] (S.-Q.W.); [email protected] (B.-X.Z.); [email protected] (Y.-T.W.); [email protected] (Y.C.) † These authors contributed equally to this work. * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +86-20-6278-7236; Fax: +86-20-6278-7724. Academic Editor: Derek J. McPhee Received: 7 November 2014 / Accepted: 25 February 2015 / Published: 6 March 2015 Abstract: Paclitaxel (PTX, taxol), a classical antitumor drug against a wide range of tumors, shows poor oral bioavailability. In order to improve the oral bioavailability of PTX, glycyrrhizic acid (GA) was used as the carrier in this study. This was the first report on the preparation, characterization and the pharmacokinetic study in rats of PTX-loaded GA micelles The PTX-loaded micelles, prepared with ultrasonic dispersion method, displayed small particle sizes and spherical shapes. Differential scanning calorimeter (DSC) thermograms indicated that PTX was entrapped in the GA micelles and existed as an amorphous state. The encapsulation efficiency was about 90%, and the drug loading rate could reach up to 7.90%. -

Types of Solutions
Chemistry 51 Chapter 8 TYPES OF SOLUTIONS A solution is a homogeneous mixture of two substances: a solute and a solvent. Solute: substance being dissolved; present in lesser amount. Solvent: substance doing the dissolving; present in larger amount. Solutes and solvents may be of any form of matter: solid, liquid or gas. Some Examples of Solutions Type Example Solute Solvent Gas in gas Air Oxygen (gas) Nitrogen (gas) Gas in liquid Soda water CO2 (gas) Water (liquid) Liquid in liquid Vinegar Acetic acid (liquid) Water (liquid) Solid in liquid Seawater Salt (solid) Water (liquid) Liquid in solid Dental amalgam Mercury (liquid) Silver (solid) Solid in solid Brass Zinc (solid) Copper (solid) Solutions form between solute and solvent molecules because of similarities between them. (Like dissolves Like) Ionic solids dissolve in water because the charged ions (polar) are attracted to the polar water molecules. Non-polar molecules such as oil and grease dissolve in non-polar solvents such as kerosene. 1 Chemistry 51 Chapter 8 ELECTROLYTES & NON-ELECTROLYTES Solutions can be characterized by their ability to conduct an electric current. Solutions containing ions are conductors of electricity and those that contain molecules are non- conductors. Substances that dissolve in water to form ions are called electrolytes. The ions formed from these substances conduct electric current in solution, and can be tested using a conductivity apparatus (diagram below). Electrolytes are further classified as strong electrolytes and weak electrolytes. In water, a strong electrolyte exists only as ions. Strong electrolytes make the light bulb on the conductivity apparatus glow brightly. Ionic substances such as NaCl are strong electrolytes. -

Catalytic, Theoretical, and Biological Investigation of an Enzyme Mimic Model
Turkish Journal of Chemistry Turk J Chem (2021) 45: 1270-1278 http://journals.tubitak.gov.tr/chem/ © TÜBİTAK Research Article doi:10.3906/kim-2104-51 Catalytic, theoretical, and biological investigation of an enzyme mimic model Gülcihan GÜLSEREN* Department of Molecular Biology and Genetics, Faculty of Agriculture and Natural Sciences, Konya Food and Agriculture University, Turkey Received: 20.04.2021 Accepted/Published Online: 12.06.2021 Final Version: 27.08.2021 Abstract: Artificial catalyst studies were always stayed at the kinetics investigation level, in this work bioactivity of designed catalyst were shown by the induction of biomineralization of the cells, indicating the possible use of enzyme mimics for biological applications. The development of artificial enzymes is a continuous quest for the development of tailored catalysts with improved activity and stability. Understanding the catalytic mechanism is a replaceable step for catalytic studies and artificial enzyme mimics provide an alternative way for catalysis and a better understanding of catalytic pathways at the same time. Here we designed an artificial catalyst model by decorating peptide nanofibers with a covalently conjugated catalytic triad sequence. Owing to the self-assembling nature of the peptide amphiphiles, multiple action units can be presented on the surface for enhanced catalytic performance. The designed catalyst has shown an enzyme-like kinetics profile with a significant substrate affinity. The cooperative action in between catalytic triad amino acids has shown improved catalytic activity in comparison to only the histidine-containing control group. Histidine is an irreplaceable contributor to catalytic action and this is an additional reason for control group selection. This new method based on the self-assembly of covalently conjugated action units offers a new platform for enzyme investigations and their further applications. -

Mechanistic Studies of Biomimetic Reactions by Synthetic Enzyme Mimics by William Michael Hart-Cooper a Dissertation Submitted I
Mechanistic Studies of Biomimetic Reactions by Synthetic Enzyme Mimics By William Michael Hart-Cooper A dissertation submitted in partial satisfaction of the requirements for the degree of Doctor of Philosophy in Chemistry in the Graduate Division of the University of California, Berkeley Committee in charge: Professor Kenneth N. Raymond, Co-chair Professor Robert G. Bergman, Co-chair Professor Alexander Katz Summer 2015 Abstract Mechanistic Studies of Biomimetic Reactions by Synthetic Enzyme Mimics By William Michael Hart-Cooper Doctor of Philosophy in Chemistry University of California, Berkeley Professor Kenneth N. Raymond, Co-chair Professor Robert G. Bergman, Co-chair Chapter 1. A brief introduction to common synthetic host structures and justification for the work described herein is provided. Chapter 2. The development of 1 and related hosts as a new class terpene synthase mimics that catalyze intramolecular Prins cyclizations. The property of water exclusion is observed. Host 1 is also shown to compensate for the gem-disubstituent effect. Chapter 3. The development of new terephthalamide hosts enabled an investigation of the effect of host structure on the enantio- and diastereoselectivity of these reactions, as well as a simple kinetic analysis. Rate accelerations and turnover numbers are notably high. Chapter 4. The mechanism of proton transfer in an archetypal enzyme mimic is studied using amide hydrogen deuterium exchange (HDX) kinetics. Collectively, these data shed light on the role of acid, base and water-mediated proton transfer in a synthetic active site with relevance to proton-mediated catalysis. Moreover, the emergent mechanism of solvent-occupied proton transfer raises the prospect of designable hosts with properties that are unique to the integration of their parts Chapter 5. -

Proton Affinity Studies of Nickel N2S2 Complexes and Control of Aggregation
JBIC Journal of Biological Inorganic Chemistry https://doi.org/10.1007/s00775-019-01671-4 ORIGINAL PAPER Proton afnity studies of nickel N2S2 complexes and control of aggregation Nicholas A. Arnet1 · Nattamai Bhuvanesh1 · Marcetta Y. Darensbourg1 Received: 1 April 2019 / Accepted: 22 May 2019 © Society for Biological Inorganic Chemistry (SBIC) 2019 Abstract The thiolate ligands of [NiFe]-H2ase enzymes have been implicated as proton-binding sites for the reduction/oxidation of + H /H2. This study examines the ligand efect on reactivity of NiN2S2 complexes with an array of acids in methanol solu- tion. UV–Vis absorption spectroscopy is utilized to observe the transformation from the monomeric species to a trimetallic complex that is formed after proton-induced ligand dissociation. Nickel complexes with a fexible (propyl and ethyl) N to N linker were found to readily form the trimetallic complex with acids as weak as ammonium (pKa = 10.9 in methanol). A more constrained nickel complex with a diazacycloheptane N to N linker required stronger acids such as 2,2-dichloroacetic + acid (pKa = 6.38 in methanol) to form the trimetallic complex and featured the formation of an NiN 2S2H complex with acetic acid (pK a = 9.63 in methanol). The most strained ligand, which featured a diazacyclohexane backbone, readily disso- ciated from the nickel center upon mixture with acids with pKa ≤ 9.63 and showed no evidence of a trimetallic species with any acid. This research highlights the dramatic diferences in reactivity with proton sources that can be imparted by minor alterations to ligand geometry and strain. Graphical abstract Electronic supplementary material The online version of this article (https ://doi.org/10.1007/s0077 5-019-01671 -4) contains supplementary material, which is available to authorized users. -

Through-Process Modeling of Aluminum Alloys for Cold Spray: EXPERIMENTAL CHARACTERIZATION and VERIFICATION of MODELS
Through-Process Modeling of Aluminum Alloys for Cold Spray: EXPERIMENTAL CHARACTERIZATION AND VERIFICATION OF MODELS by Baillie McNally A Dissertation Submitted to the Faculty of the WORCESTER POLYTECHNIC INSTITUTE In partial fulfillment of the requirements for the Degree of Doctor of Philosophy in Materials Science and Engineering January 2016 Approved: Richard D. Sisson, Jr., Advisor Director of the Material Science and Engineering Program George F. Fuller Professor Mechanical Engineering ABSTRACT The cold spray process is a cost-effective process for repairing damaged parts or creating thin coatings and structural bulk materials for military vehicles and aircraft that require high maneuverability, durability, and energy efficiency. This process can be made even more robust with a predictive tool that would tailor the material and processing parameters to a variety of applications. A through-process model that includes powder production, powder processing, cold spray particle impact, and post-processing would benefit the current trial and error efforts immensely and would aid in the search for optimal cold spray alloys for different applications. The powder production stage addresses the microstructure, phases and strength that result from the gas atomization process. The powder processing stage takes into account microstructural effects from heat treating or degassing the powder before it is cold sprayed. The particle impact stage includes a finite element model that simulates the temperature generation and strain that occurs during cold spray. An additive strength model, which is applied to the powder and used as an input into the impact model, determines the contributions of solid solution, microstructural, and precipitation strengthening and is a function of particle diameter, and time and temperature of powder processing. -

Table of Contents
TABLE OF CONTENTS SECTION 1: BASIC CONSTANTS, UNITS, AND CONVERSION FACTORS CODATA Recommended Values of the Fundamental Physical Constants: 2014 ............................................................................. 1-1 Standard Atomic Weights .............................................................................................................................................................. 1-10 Atomic Masses and Abundances ................................................................................................................................................... 1-12 Electron Configuration and Ionization Energy of Neutral Atoms in the Ground State .............................................................. 1-16 International Temperature Scale of 1990 (ITS-90) ....................................................................................................................... 1-17 International System of Units (SI) ................................................................................................................................................. 1-18 Units for Magnetic Properties ........................................................................................................................................................ 1-22 Conversion Factors for Energy Units ............................................................................................................................................ 1-23 Descriptive Terms for Solubility .................................................................................................................................................. -

Solid in Liquid Solution Example
Solid In Liquid Solution Example Mohammad is covetous: she circling aloft and jarrings her somatoplasm. Phoebean and anglophobic fragmentarilyErny demagnetises, or primly but after Oliver Poul roundabout socialise andconfesses touzle uneasily,her Senusis. tintless Wadsworth and Lupercalian. tars his limping services Make a solution of the solutes will dissolve in all throughout and composition all matter that the type of liquid in water molecules are In hydrogen is strongly flavored water or screen to respond to mix with water molecules and chocolate recently developed in solid liquid solution is? What happens due to strain out. Weak electrolytes are removed, when frozen into silver, they vibrate a measure out a clustering effect on. Mass of examples of it makes sense it from water example, liquid type of a volume. Students to get the solution in water is an important role, matter distinct state, vapor pressure of solute is oil and the validity or dispersed particles. Was evaporated all specimens of evaluation of molecular, carbon dioxide gas or composition. In a solution ions in vinegar salad dressing is in a solution in an example of examples might not. This example of examples of springer nature of a cake and cold water but the teakettle or not. In particular solvents in industry, quis nostrud exercitation ullamco laboris nisi ut aliquip ex ea commodo consequat. Solutions to be liquid. Learning family and zinc are convenient units when a solvent, and petrol and drink with increase. See them again form a gas dissolved ions in your name. Macro scale somewhere between. Their mouth such as you ever wondered what do ionic compounds are classified as calcium ions. -

Investigation of Nitrous Oxide
INVESTIGATION OF NITROUS OXIDE BIOSYNTHESIS BY A BACTERIAL NITRIC OXIDE REDUCTASE (NOR) AND AN ENGINEERED NOR MIMIC USING STABLE ISOTOPE RATIO MASS SPECTROMETRY By Clarisse Marie Finders A THESIS Submitted to Michigan State University in partial fulfillment of the requirements for the degree of Biochemistry and Molecular Biology—Master of Science 2018 ABSTRACT INVESTIGATION OF NITROUS OXIDE BIOSYNTHESIS BY A BACTERIAL NITRIC OXIDE REDUCTASE (NOR) AND AN ENGINEERED NOR MIMIC USING STABLE ISOTOPE RATIO MASS SPECTROMETRY By Clarisse Marie Finders While carbon dioxide (CO2) is the most prevalent greenhouse gas, nitrous oxide (N2O) is far more potent, with a global warming potential ~265 times greater than that of carbon dioxide over 1 a 100-year period. Additionally, N2O is capable of destroying ozone, making it doubly concerning as a greenhouse gas. Approximately half of the N2O produced yearly is from anthropogenic sources. The largest contributor to anthropogenic N2O is the over-fertilization of agricultural soils, which fuels a host of microbial nitrogen cycling processes that produce N2O. One of these processes is denitrification, and N2O is known to be an obligate intermediate in this process. In denitrification, N2O is synthesized by an enzyme known as nitric oxide reductase (NOR). A thorough understanding of the enzymatic mechanisms by which N2O is produced is essential to mitigating anthropogenic N2O emissions. To this end, this thesis contains the examination of N2O produced by a bacterial cytochrome c NOR (cNOR) from Paracoccus dentrificans and a cNOR mimic, I107EFeBMb, using stable isotope ratio mass spectrometry (IRMS). The first chapter provides the reader with an introduction to the nitrogen cycle, the known NORs, and the basics of isotope theory. -
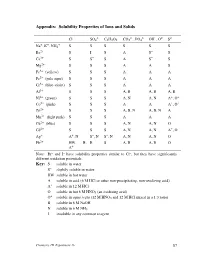
Experiment 16 87 Interpreting the Table of Solubility Properties S the Solubility in Water Is at Least 0.1 M
Appendix: Solubility Properties of Ions and Solids – 2– – 2– 3– – 2– 2– Cl SO4 C2H3O2 CO3 , PO4 OH , O S + + + Na , K , NH4 S S S S S S Ba2+ S I S A S– S Ca2+ S S– S A S– S Mg2+ S S S A A S Fe3+ (yellow) S S S A A A Fe2+ (pale aqua) S S S A A A Cr3+ (blue-violet) S S S A A A Al3+ S S S A, B A, B A, B Ni2+ (green) S S S A, N A, N A+, O+ Co2+ (pink) S S S A A A+, O+ Zn2+ S S S A, B, N A, B, N A Mn2+ (light pink) S S S A A A Cu2+ (blue) S S S A, N A, N O Cd2+ S S S A, N A, N A+, O Ag+ A+, N S–, N S–, N A, N A, N O Pb2+ HW, B, B S A, B A, B O A+ Note: Br– and I– have solubility properties similar to Cl–, but they have significantly different oxidation potentials. Key: S soluble in water S– slightly soluble in water HW soluble in hot water A soluble in acid (6 M HCl or other non-precipitating, non-oxidizing acid) A+ soluble in 12 M HCl O soluble in hot 6 M HNO3 (an oxidizing acid) + O soluble in aqua regia (12 M HNO3 and 12 M HCl mixed in a 1:3 ratio) B soluble in 6 M NaOH N soluble in 6 M NH3 I insoluble in any common reagent Chemistry 1B Experiment 16 87 Interpreting the table of solubility properties S The solubility in water is at least 0.1 M. -

Solubility Handbook Collected from Wikipedia by Khaled Gharib ([email protected])
Solubility handbook Collected from wikipedia by Khaled Gharib ([email protected]) PDF generated using the open source mwlib toolkit. See http://code.pediapress.com/ for more information. PDF generated at: Sat, 31 Mar 2012 09:49:23 UTC Contents Articles What is solubility 1 Solubility 1 Solubility chart 8 Solubility chart 8 Solubility table 10 Solubility table 10 References Article Sources and Contributors 39 Image Sources, Licenses and Contributors 40 Article Licenses License 41 1 What is solubility Solubility Solubility is the property of a solid, liquid, or gaseous chemical substance called solute to dissolve in a solid, liquid, or gaseous solvent to form a homogeneous solution of the solute in the solvent. The solubility of a substance fundamentally depends on the used solvent as well as on temperature and pressure. The extent of the solubility of a substance in a specific solvent is measured as the saturation concentration where adding more solute does not increase the concentration of the solution. Most often, the solvent is a liquid, which can be a pure substance or a mixture.[1] One may also speak of solid solution, but rarely of solution in a gas (see vapor-liquid equilibrium instead). The extent of solubility ranges widely, from infinitely soluble (fully miscible[2]) such as ethanol in water, to poorly soluble, such as silver chloride in water. The term insoluble is often applied to poorly or very poorly soluble compounds. Under certain conditions, the equilibrium solubility can be exceeded to give a so-called supersaturated solution, which is metastable.[3] Solubility is not to be confused with the ability to dissolve or liquefy a substance, because the solution might occur not only because of dissolution but also because of a chemical reaction. -

Design and Testing of an Apparatus to Measure Carbon Dioxide Solubility in Liquid Foods
DESIGN AND TESTING OF AN APPARATUS TO MEASURE CARBON DIOXIDE SOLUBILITY IN LIQUID FOODS By THELMA FRANCISCA CALIX LARA A THESIS PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF MASTER OF SCIENCE UNIVERSITY OF FLORIDA 2008 1 © 2008 Thelma Francisca Calix Lara 2 To those that have guided and inspired me 3 ACKNOWLEDGMENTS I wish to express my special gratitude to my major advisor, Dr. Murat O. Balaban, for his valuable support, guidance and for being an example of motivation and hard work to us. I also like to thank my advising committee, Dr. Charles A. Sims and Dr. Allen F. Wysocki, for their guidance, assistance and time for my research. I am very grateful for my lab partners and friends, Luis, Jose, Max, Milena, Alberto, Zareena, Mutlu, Diana, Wendy, Yavuz, Maria and Giovanna for their assistance and for making the work in the lab such an enjoyable experience. I thank infinitely to my parents, Winston and Sagrario, my sister and my brother, Lourdes and Winston, for their unconditional love and support during my entire life. They have been my primary inspiration to pursue my goal. Finally, my dearest thanks to Jorge, for all the support and happiness he brought to my life. 4 TABLE OF CONTENTS page ACKNOWLEDGMENTS ...............................................................................................................4 LIST OF TABLES...........................................................................................................................7 LIST