Tools for Maximizing the Efficiency of Protein
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Dr. Martin St. Maurice's Publications
Dr. Martin St. Maurice’s Publications 2013 Lin, Y., and St. Maurice, M. 2013. The structure of allophanate hydrolase from Granulibacter bethesdensis provides insights into substrate specificity in the amidase signature family. Biochemistry. 52: 690-700. 2012 Waldrop, G.L., Holden, H.M., and St. Maurice, M. 2012. The enzymes of biotin dependent CO2 metabolism: What structures reveal about their reaction mechanisms. Protein Science 21(11):1597-1619. Adina-Zada, A., Sereeruk, C., Jitrapakdee, S., Zeczycki, T.N., St. Maurice, M., Cleland, W.W., Wallace, J.C., and Attwood, P.V. 2012. Roles of Arg427 and Arg472 in the binding and allosteric effects of acetyl CoA in pyruvate carboxylase. Biochemistry 51(41): 1597-1619. 2011 Adina-Zada, A., Hazra, R., Sereeruk, C., Jitrapakdee, S., Zeczycki, T.N., St. Maurice, M., Cleland, W.W., Wallace, J.C., and Attwood, P.V. 2011. Probing the allosteric activation of pyruvate carboxylase using 2′,3′-O-(2,4,6-trinitrophenyl) adenosine 5′-triphosphate as a fluorescent mimic of the allosteric activator acetyl CoA. Arch. Biochem. Biophys. 117-126. Zeczycki, T.N., Menefee, A.L., Jitrapakdee, S., Wallace, J.C., Attwood, P.V., St. Maurice, M. and Cleland, W.W. 2011. Activation and inhibition of pyruvate carboxylase from Rhizobium etli. Biochemistry. 9694-9707. Lietzan, A.D., Menefee, A.L., Zeczycki, T.N., Kumar, S., Attwood, P.V., Wallace, J.C., Cleland, W.W. and St. Maurice, M. 2011. Interaction between the biotin carrier domain and the biotin carboxylase domain in the structure of Rhizobium etli pyruvate carboxylase. Biochemistry. 9708-9723. Zeczycki, T.N., Menefee, A.L., Adina-Zada, A., Surinya, K.H., Wallace, J.C., Attwood, P.V., St. -

Functional Assignments in the Enolase Superfamily: Investigations of Two Divergent Groups of D-Galacturonate Dehydratases and Galactarate Dehydratase-Iii
FUNCTIONAL ASSIGNMENTS IN THE ENOLASE SUPERFAMILY: INVESTIGATIONS OF TWO DIVERGENT GROUPS OF D-GALACTURONATE DEHYDRATASES AND GALACTARATE DEHYDRATASE-III BY FIONA PATRICIA GRONINGER-POE i DISSERTATION Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biochemistry in the Graduate College of the University of Illinois at Urbana-Champaign, 2014 Urbana, Illinois Doctoral Committee: Professor John A. Gerlt, Chair Professor John E. Cronan, Jr. Associate Professor Rutilo Fratti Associate Professor Raven H. Huang ABSTRACT More than a decade after the genomic age, full genome sequencing is cost-effective and fast, allowing for the deposit of an ever increasing number of DNA sequences. New fields have arisen from this availability of genomic information, and the way we think about biochemistry and enzymology has been transformed. Unfortunately, there is no robust method for accurately determining the functions of enzymes encoded by these sequences that matches the speed in which genomes are deposited into public databases. Functional assignment of enzymes remains of utmost importance in understanding microbial metabolism and has applications in agriculture by examining bacterial plant pathogen metabolism and additionally in human health by providing metabolic context to the human gut microbiome. To aid in the functional identification of proteins, enzymes can be grouped into superfamilies which share common structural motifs as well as mechanistic features. To this end, the enolase superfamily is an excellent model system for functional assignment because more than half of the members still lack functional identification. Structurally, these enzymes contain substrate specificity residues in the N-terminal capping domain and catalytic residues in the C-terminal barrel domain. -

The Structure of Neurospora Crassa 3-Carboxy-Cis,Cis-Muconate Lactonizing Enzyme, a  Propeller Cycloisomerase
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Structure, Vol. 10, 483–492, April, 2002, 2002 Elsevier Science Ltd. All rights reserved. PII S0969-2126(02)00744-X The Structure of Neurospora crassa 3-Carboxy-cis,cis-Muconate Lactonizing Enzyme, a  Propeller Cycloisomerase Tommi Kajander,1 Michael C. Merckel,1 ing the structural basis for different alternatives for catal- Andrew Thompson,2 Ashley M. Deacon,3 ysis in MLEs could therefore aid in understanding and Paul Mazur,4 John W. Kozarich,4,6 engineering enzymes for degradation of xenobiotic pol- and Adrian Goldman1,5 lutants, such as fluoroaromatics. 1 Institute of Biotechnology MLEs convert cis,cis-muconates to muconolactones Research Program in Structural Biology in soil microbes as part of the -ketoadipate pathway. and Biophysics This aerobic catabolic pathway converts aromatics, such University of Helsinki as the breakdown products of lignin, through catechol FIN-00014 Helsinki and protocatechuate to citric acid cycle intermediates. Finland The two major branches of the -ketoadipate pathway 2 EMBL are the catechol branch, with cis,cis-muconate lactoniz- Grenoble Outstation ing enzymes, and the protocatechuate branch (Figure 38024 Grenoble 1), with carboxy-cis,cis-muconate lactonizing enzymes France (CMLEs). 3 Stanford Synchrotron Radiation Laboratory The evolutionary origins of these enzymes are surpris- Menlo Park, California 94025 ingly divergent, both in terms of sequence similarity and 4 Merck Research Laboratories functional properties (Table 1). Three different classes of Rahway, New Jersey 07065 lactonizing enzymes can be distinguished. The bacterial MLEs contain an unusual TIM barrel [3] and are closely related to mandelate racemase [4]. -

SI Appendix Index 1
SI Appendix Index Calculating chemical attributes using EC-BLAST ................................................................................ 2 Chemical attributes in isomerase reactions ............................................................................................ 3 Bond changes …..................................................................................................................................... 3 Reaction centres …................................................................................................................................. 5 Substrates and products …..................................................................................................................... 6 Comparative analysis …........................................................................................................................ 7 Racemases and epimerases (EC 5.1) ….................................................................................................. 7 Intramolecular oxidoreductases (EC 5.3) …........................................................................................... 8 Intramolecular transferases (EC 5.4) ….................................................................................................. 9 Supporting references …....................................................................................................................... 10 Fig. S1. Overview …............................................................................................................................ -

Functional and Physiological Discovery in the Mannonate Dehydratase Subgroup of the Enolase Superfamily
FUNCTIONAL AND PHYSIOLOGICAL DISCOVERY IN THE MANNONATE DEHYDRATASE SUBGROUP OF THE ENOLASE SUPERFAMILY BY DANIEL JOSEPH WICHELECKI DISSERTATION Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biochemistry in the Graduate College of the University of Illinois at Urbana-Champaign, 2014 Urbana, Illinois Doctoral Committee: Professor John Gerlt, Chair Professor John Cronan Professor Scott Silverman Professor Wilfred van der Donk ABSTRACT In the current post-genomic world, the exponential amassing of protein sequences is overwhelming the scientific community’s ability to experimentally assign each protein’s function. The use of automated, homology-based annotations has allowed a reprieve from this efflux of data, but has led to widespread misannotation and nonannotation in protein sequence databases. This dissertation details the functional and physiological characterization of the mannonate dehydratase subgroup (ManD) of the enolase superfamily (ENS). The outcome affirms the dangers of homology-based annotations while discovering novel metabolic pathways. Furthermore, the experimental verification of these pathways ( in vitro and in vivo ) has provided a platform to test the general strategies for improved functional and metabolic characterization being developed by the Enzyme Function Initiative (EFI). Prior to this study, one member of the ManD subgroup had been characterized and was shown to dehydrate D-mannonate to 2-keto-3-deoxy-D-gluconate. Forty-two additional members of the ManD, selected from across the sequence space of the subgroup, were screened for activity and kinetic constants were determined. The members of the once isofunctional subgroup were found to differ in both catalytic efficiency and substrate specificity: 1) high 3 4 -1 -1 efficiency (k cat /K M = 10 to 10 M s ) dehydration of D-mannonate, 2) low efficiency (k cat /K M = 10 1 to 10 2 M-1s-1) dehydration of D-mannonate and/or D-gluconate, and 3) no-activity with either D-mannonate or D-gluconate (or any other acid sugar tested). -

Supporting Information for Proteomics DOI 10.1002/Pmic.200600987
Supporting Information for Proteomics DOI 10.1002/pmic.200600987 Lars Whlbrand, Birte Kallerhoff, Daniela Lange, Peter Hufnagel, Jrgen Thiermann, Richard Reinhardt and Ralf Rabus Functional proteomic view of metabolic regulation in “Aromatoleum aromaticum” strain EbN1 ª 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com Content 1. General proteome features Page Experimental outline of the presented study with “Aromatoleum aromaticum” 1-2 strain EbN1 (Fig. 1.1.) Spot pattern comparison of differently visualized 2DE gels (Fig. 1.2.) 1-3 Principle component analysis of the proteins for each analyzed 2D DIGE gel 1-4 of all main physiological groups (Fig. 1.3.) Fold changes in protein abundance of all identified proteins (Fig 1-5 Number of regulated as well as not regulated protein spots on the 2DE gels 1-6 and their share of total protein (Table 1.1.) Mean average ratio of not regulated protein spots of the three main 1-7 physiological groups (Table 1.2.) Mean average ratios of not regulated proteins, identified in all three main 1-8 physiological groups (Table 1.3.) Proteins identified in this study (Table 1.4.) 1-9 2. Cellular functions* 2.1. DNA replication, recombination and repair 2-2 2.1.1. DNA replication 2-2 2.2. Transcription 2-4 2.2.1. DNA-dependent RNA polymerase and sigma factors 2-5 2.2.2. Factors affecting RNA polymerase 2-5 2.2.3. Classes of transcriptional regulators 2-5 2.3. Translation 2-6 2.3.1. tRNA synthesis 2-6 2.3.2. tRNA/rRNA modification 2-7 2.3.3. -

Oup Proeng Gzy011 135..145 ++
Protein Engineering, Design & Selection, 2018, vol. 31 no. 4, pp. 135–145 doi: 10.1093/protein/gzy011 Advance Access Publication Date: 30 May 2018 Original Article Original Article A platform for chemical modification of mandelate racemase: characterization of the C92S/C264S and γ-thialysine 166 variants Downloaded from https://academic.oup.com/peds/article/31/4/135/5025590 by guest on 30 September 2021 Mitesh Nagar1, Himank Kumar1, and Stephen L. Bearne1,2,* 1Department of Biochemistry and Molecular Biology, Dalhousie University, Halifax, NS, Canada B3H 4R2, and 2Department of Chemistry, Dalhousie University, Halifax, NS, Canada B3H 4R2 *To whom correspondence should be addressed. E-mail: [email protected] Edited by Miroslaw Cygler Received 7 February 2018; Revised 28 April 2018; Editorial Decision 30 April 2018; Accepted 3 May 2018 Abstract Mandelate racemase (MR) serves as a paradigm for our understanding of enzyme-catalyzed deprotona- tion of a carbon acid substrate. To facilitate structure–function studies on MR using non-natural amino acid substitutions, we engineered the Cys92Ser/Cys264Ser variant (dmMR) as a platform for introdu- cingCysresiduesatspecific locations for subsequent covalent modification. While the highly reactive thiol of Cys furnishes a site for chemical modification, site-specificity requires that other Cys residues be non-reactive or replaced by a non-reactive amino acid, especially if chemical modification is con- ducted under denaturing conditions. The catalytic efficiency of dmMR is reduced only ~2-fold relative to wild-type MR, making dmMR a viable platform for the site-specificintroductionofCys.Asan example, the inactive Lys166Cys variant of dmMR was treated with ethylenimine under denaturing con- ditions to replace the Brønsted acid-base catalyst Lys 166 with the non-natural amino acid γ-thialysine. -

Impact of Temperature on the Time Required for the Establishment of Primordial Biochemistry, and for the Evolution of Enzymes
Impact of temperature on the time required for the establishment of primordial biochemistry, and for the evolution of enzymes Randy B. Stockbridge, Charles A. Lewis, Jr., Yang Yuan, and Richard Wolfenden1 Department of Biochemistry and Biophysics, University of North Carolina, Chapel Hill, NC 27599 Contributed by Richard Wolfenden, September 29, 2010 (sent for review August 1, 2010) All reactions are accelerated by an increase in temperature, but the magnitude of that effect on very slow reactions does not seem to have been fully appreciated. The hydrolysis of polysaccharides, for example, is accelerated 190,000-fold when the temperature is raised from 25 to 100 °C, while the rate of hydrolysis of phosphate monoester dianions increases 10,300,000-fold. Moreover, the slow- est reactions tend to be the most heat-sensitive. These tendencies collapse, by as many as five orders of magnitude, the time that would have been required for early chemical evolution in a warm environment. We propose, further, that if the catalytic effect of a “proto-enzyme”—like that of modern enzymes—were mainly enthalpic, then the resulting rate enhancement would have in- creased automatically as the environment became cooler. Several powerful nonenzymatic catalysts of very slow biological reactions, notably pyridoxal phosphate and the ceric ion, are shown to meet that criterion. Taken together, these findings greatly reduce the time that would have been required for early chemical evolution, countering the view that not enough time has passed for life to Fig. 1. Half-lives (t1∕2) and first order rate constants (k) of some biological have evolved to its present level of complexity. -

Crystal Structure Analysis of a Bacterial Lysozyme at Very High Resolution
The crystal structure of a bacterial lysozyme at atomic resolution Dissertation zur Erlangung des akademischen Grades doctor rerum naturalium (Dr. rer. nat.) vorgelegt dem Rat der Chemisch-Geowissenschaftlichen Fakultät der Friedrich-Schiller-Universität Jena von Diplom-Chemikerin Astrid Rau geboren am 01.06.1974 in Saalfeld Gutachter: 1. Prof. Dr. R. Hilgenfeld 2. Prof. Dr. D. Klemm Tag der öffentlichen Verteidigung: 01.06.2005 TABLE OF CONTENTS 1. INTRODUCTION 1 1.1 Milestones in lysozyme research 1 1.2 Definition and classification of lysozymes 5 1.3 Catalytic mechanisms of lysozymes 7 1.4 Chalaropsis-type lysozymes 9 1.5 Cellosyl – a Ch-type lysozyme from Streptomyces coelicolor 12 1.7 Aim of the project 13 2. MATERIALS AND METHODS 14 2.1 Materials 14 2.1.1 Proteins 14 2.1.2 Carbohydrates 14 2.1.3 Chemicals 15 2.1.4 Crystallisation screens 15 2.1.5 Dialysing tools, assays, crystallisation materials and cryo-tools 15 2.1.6 Laboratory equipment and synchrotron facilities 16 2.2 Methods 17 2.2.1 Determination of protein purity 17 2.2.2 Determination of protein concentration 18 2.2.3 Dialysis 18 2.2.4 Sample concentration 18 2.2.5 Crystallisation 18 2.2.6 Heavy atom and polysaccharide soaks 19 2.2.7 Cryocooling 20 2.2.8 Data acquisition and processing 20 2.2.8.1 Native data collection on the monoclinic crystal form 21 2.2.8.2 Native data collection on the hexagonal crystal form 22 2.2.8.3 MAD data collection 22 2.2.8.4 Data collection on heavy-atom derivatised crystals 24 2.2.8.5 Collection and processing of atomic-resolution data 25 I 2.2.9 Phase determination 27 2.2.9.1 Molecular replacement 28 2.2.9.2 Multiple wavelength anomalous dispersion 29 2.2.9.3 Multiple isomorphous replacement with anomalous scattering 29 2.2.10 Model building and electron-density maps 31 2.2.11 Structure refinement 33 2.2.12 Validation of model quality 35 3. -
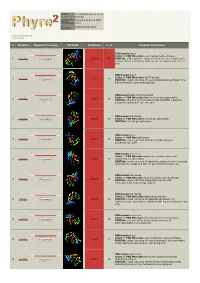
Phyre 2 Results for P29208
Email [email protected] Description P29208 Thu Jan 5 11:45:36 GMT Date 2012 Unique Job 0bd634b8f8620003 ID Detailed template information # Template Alignment Coverage 3D Model Confidence % i.d. Template Information PDB header:lyase Chain: A: PDB Molecule:o-succinylbenzoate synthase; 1 c3gc2A_ Alignment 100.0 86 PDBTitle: 1.85 angstrom crystal structure of o-succinylbenzoate synthase from2 salmonella typhimurium in complex with succinic acid PDB header:lyase Chain: A: PDB Molecule:tlr1174 protein; 2 c2oztA_ 100.0 20 Alignment PDBTitle: crystal structure of o-succinylbenzoate synthase from2 thermosynechococcus elongatus bp-1 PDB header:metal binding protein Chain: A: PDB Molecule:enzyme of enolase superfamily; 3 c3px5A_ 100.0 19 Alignment PDBTitle: structure of efi enolase target en500555, a putative dipeptide2 epimerase: apo structure PDB header:isomerase 4 c1jpmB_ Alignment 100.0 20 Chain: B: PDB Molecule:l-ala-d/l-glu epimerase; PDBTitle: l-ala-d/l-glu epimerase PDB header:lyase Chain: A: PDB Molecule:menc; 5 c2pgeA_ 100.0 23 Alignment PDBTitle: crystal structure of menc from desulfotalea psychrophila2 lsv54 PDB header:isomerase Chain: E: PDB Molecule:mandelate racemase/muconate 6 c3q45E_ Alignment 100.0 19 lactonizing enzyme family; PDBTitle: crystal structure of dipeptide epimerase from cytophaga hutchinsonii2 complexed with mg and dipeptide d-ala-l-val PDB header:isomerase Chain: A: PDB Molecule:chloromuconate cycloisomerase; 7 c1nu5A_ 100.0 16 Alignment PDBTitle: crystal structure of pseudomonas sp. p51 chloromuconate lactonizing2 -

The Enolase Superfamily: a General Strategy for Enzyme-Catalyzed Abstraction of the R-Protons of Carboxylic Acids†
+ + Biochemistry 1996, 35, 16489-16501 16489 The Enolase Superfamily: A General Strategy for Enzyme-Catalyzed Abstraction of the R-Protons of Carboxylic Acids† ,‡ §,| ⊥,# Patricia C. Babbitt,* Miriam S. Hasson, Joseph E. Wedekind, David R. J. Palmer,r William C. Barrett,r ⊥ ⊥ § ‡ , George H. Reed, Ivan Rayment, Dagmar Ringe, George L. Kenyon, and John A. Gerlt* r Department of Pharmaceutical Chemistry, UniVersity of California, San Francisco, California 94143-0446, Departments of Biochemistry and Chemistry and Rosenstiel Center for Basic Biomedical Research, Brandeis UniVersity, Waltham, Massachusetts 02154-9110, The Institute for Enzyme Research and Department of Biochemistry, UniVersity of Wisconsin, Madison, Wisconsin 53706, and Department of Biochemistry, UniVersity of Illinois, Urbana, Illinois 61801 ReceiVed July 5, 1996X ABSTRACT: We have discovered a superfamily of enzymes related by their ability to catalyze the abstraction of the R-proton of a carboxylic acid to form an enolic intermediate. Although each reaction catalyzed by these enzymes is initiated by this common step, their overall reactions (including racemization, â-elimination of water, â-elimination of ammonia, and cycloisomerization) as well as the stereochemical consequences (syn Vs anti) of the â-elimination reactions are diverse. Analysis of sequence and structural similarities among these proteins suggests that all of their chemical reactions are mediated by a common active site architecture modified through evolution to allow the enolic intermediates to partition to different products in their respective active sites Via different overall mechanisms. All of these enzymes retain the ability to catalyze the thermodynamically difficult step of proton abstraction. These homologous proteins, designated the “enolase superfamily”, include enolase as well as more metabolically specialized enzymes: mandelate racemase, galactonate dehydratase, glucarate dehydratase, muconate-lactonizing enzymes, N-acylamino acid racemase, â-methylaspartate ammonia-lyase, and o-succinylbenzoate synthase. -

Α-Methylacyl-Coa Racemase and Argininosuccinate Lyase
A460etukansi.fm Page 1 Friday, May 26, 2006 11:40 AM A 460 OULU 2006 A 460 UNIVERSITY OF OULU P.O. Box 7500 FI-90014 UNIVERSITY OF OULU FINLAND ACTA UNIVERSITATIS OULUENSIS ACTA UNIVERSITATIS OULUENSIS ACTA A SERIES EDITORS SCIENTIAE RERUM Prasenjit Bhaumik NATURALIUM Prasenjit Bhaumik Prasenjit ASCIENTIAE RERUM NATURALIUM Professor Mikko Siponen PROTEIN CRYSTALLOGRAPHIC BHUMANIORA STUDIES TO UNDERSTAND Professor Harri Mantila THE REACTION MECHANISM CTECHNICA Professor Juha Kostamovaara OF ENZYMES: DMEDICA α Professor Olli Vuolteenaho -METHYLACYL-CoA RACEMASE AND ESCIENTIAE RERUM SOCIALIUM Senior assistant Timo Latomaa ARGININOSUCCINATE LYASE FSCRIPTA ACADEMICA Communications Officer Elna Stjerna GOECONOMICA Senior Lecturer Seppo Eriksson EDITOR IN CHIEF Professor Olli Vuolteenaho EDITORIAL SECRETARY Publication Editor Kirsti Nurkkala FACULTY OF SCIENCE, DEPARTMENT OF BIOCHEMISTRY, BIOCENTER OULU, ISBN 951-42-8090-3 (Paperback) UNIVERSITY OF OULU ISBN 951-42-8091-1 (PDF) ISSN 0355-3191 (Print) ISSN 1796-220X (Online) ACTA UNIVERSITATIS OULUENSIS A Scientiae Rerum Naturalium 460 PRASENJIT BHAUMIK PROTEIN CRYSTALLOGRAPHIC STUDIES TO UNDERSTAND THE REACTION MECHANISM OF ENZYMES: α-METHYLACYL-COA RACEMASE AND ARGININOSUCCINATE LYASE Academic Dissertation to be presented with the assent of the Faculty of Science, University of Oulu, for public discussion in Kuusamonsali (Auditorium YB210), Linnanmaa, on June 6th, 2006, at 12 noon OULUN YLIOPISTO, OULU 2006 Copyright © 2006 Acta Univ. Oul. A 460, 2006 Supervised by Professor Rik Wierenga Reviewed