Assembly of an Active Enzyme by the Linkage of Two Protein Modules
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Dr. Martin St. Maurice's Publications
Dr. Martin St. Maurice’s Publications 2013 Lin, Y., and St. Maurice, M. 2013. The structure of allophanate hydrolase from Granulibacter bethesdensis provides insights into substrate specificity in the amidase signature family. Biochemistry. 52: 690-700. 2012 Waldrop, G.L., Holden, H.M., and St. Maurice, M. 2012. The enzymes of biotin dependent CO2 metabolism: What structures reveal about their reaction mechanisms. Protein Science 21(11):1597-1619. Adina-Zada, A., Sereeruk, C., Jitrapakdee, S., Zeczycki, T.N., St. Maurice, M., Cleland, W.W., Wallace, J.C., and Attwood, P.V. 2012. Roles of Arg427 and Arg472 in the binding and allosteric effects of acetyl CoA in pyruvate carboxylase. Biochemistry 51(41): 1597-1619. 2011 Adina-Zada, A., Hazra, R., Sereeruk, C., Jitrapakdee, S., Zeczycki, T.N., St. Maurice, M., Cleland, W.W., Wallace, J.C., and Attwood, P.V. 2011. Probing the allosteric activation of pyruvate carboxylase using 2′,3′-O-(2,4,6-trinitrophenyl) adenosine 5′-triphosphate as a fluorescent mimic of the allosteric activator acetyl CoA. Arch. Biochem. Biophys. 117-126. Zeczycki, T.N., Menefee, A.L., Jitrapakdee, S., Wallace, J.C., Attwood, P.V., St. Maurice, M. and Cleland, W.W. 2011. Activation and inhibition of pyruvate carboxylase from Rhizobium etli. Biochemistry. 9694-9707. Lietzan, A.D., Menefee, A.L., Zeczycki, T.N., Kumar, S., Attwood, P.V., Wallace, J.C., Cleland, W.W. and St. Maurice, M. 2011. Interaction between the biotin carrier domain and the biotin carboxylase domain in the structure of Rhizobium etli pyruvate carboxylase. Biochemistry. 9708-9723. Zeczycki, T.N., Menefee, A.L., Adina-Zada, A., Surinya, K.H., Wallace, J.C., Attwood, P.V., St. -

Assembly of an Active Enzyme by the Linkage of Two Protein Modules
Proc. Natl. Acad. Sci. USA Vol. 94, pp. 1069–1073, February 1997 Biochemistry Assembly of an active enzyme by the linkage of two protein modules A. E. NIXON,M.S.WARREN, AND S. J. BENKOVIC* 152 Davey Laboratory, Department of Chemistry, Pennsylvania State University, University Park, PA 16802-6300 Contributed by S. J. Benkovic, December 9, 1996 ABSTRACT The feasibility of creating new enzyme activ- design enzymes with novel properties. Previous approaches to ities from enzymes of known function has precedence in view the design of proteins with novel activities have included of protein evolution based on the concepts of molecular catalytic antibodies (8, 9); introduction of metal ion binding recruitment and exon shuffling. The enzymes encoded by the sites, such as the one engineered into trypsin to allow either Escherichia coli genes purU and purN, N10-formyltetrahydro- control of the proteolytic activity (10) or to regulate specificity folate hydrolase and glycinamide ribonucleotide (GAR) trans- (11); creation of hybrid enzymes through exchange of subunits formylase, respectively, catalyze similiar yet distinct reactions. to create hybrid oligomers (12); replacement of structural N10-formyltetrahydrofolate hydrolase uses water to cleave elements such as the DNA binding domain of GCN4 with that N10-formyltetrahydrofolate into tetrahydrofolate and for- of CyEBP (13); mutation of multiple individual residues to mate, whereas GAR transformylase catalyses the transfer of change the cofactor specificity of glutathione reductase from formyl from N10-formyltetrahydrofolate to GAR to yield NADPH to NADH (14), modulation of the substrate speci- formyl-GAR and tetrahydrofolate. The two enzymes show ficity of aspartate aminotransferase (15); and changing the significant homology ('60%) in the carboxyl-terminal region specificity of subtilisin (16) and a-lytic protease (17) through which, from the GAR transformylase crystal structure and mutation of single functional groups. -

Resolution of Carbon Metabolism and Sulfur-Oxidation Pathways of Metallosphaera Cuprina Ar-4 Via Comparative Proteomics
JOURNAL OF PROTEOMICS 109 (2014) 276– 289 Available online at www.sciencedirect.com ScienceDirect www.elsevier.com/locate/jprot Resolution of carbon metabolism and sulfur-oxidation pathways of Metallosphaera cuprina Ar-4 via comparative proteomics Cheng-Ying Jianga, Li-Jun Liua, Xu Guoa, Xiao-Yan Youa, Shuang-Jiang Liua,c,⁎, Ansgar Poetschb,⁎⁎ aState Key Laboratory of Microbial Resources, Institute of Microbiology, Chinese Academy of Sciences, Beijing, PR China bPlant Biochemistry, Ruhr University Bochum, Bochum, Germany cEnvrionmental Microbiology and Biotechnology Research Center, Institute of Microbiology, Chinese Academy of Sciences, Beijing, PR China ARTICLE INFO ABSTRACT Article history: Metallosphaera cuprina is able to grow either heterotrophically on organics or autotrophically Received 16 March 2014 on CO2 with reduced sulfur compounds as electron donor. These traits endowed the species Accepted 6 July 2014 desirable for application in biomining. In order to obtain a global overview of physiological Available online 14 July 2014 adaptations on the proteome level, proteomes of cytoplasmic and membrane fractions from cells grown autotrophically on CO2 plus sulfur or heterotrophically on yeast extract Keywords: were compared. 169 proteins were found to change their abundance depending on growth Quantitative proteomics condition. The proteins with increased abundance under autotrophic growth displayed Bioleaching candidate enzymes/proteins of M. cuprina for fixing CO2 through the previously identified Autotrophy 3-hydroxypropionate/4-hydroxybutyrate cycle and for oxidizing elemental sulfur as energy Heterotrophy source. The main enzymes/proteins involved in semi- and non-phosphorylating Entner– Industrial microbiology Doudoroff (ED) pathway and TCA cycle were less abundant under autotrophic growth. Also Extremophile some transporter proteins and proteins of amino acid metabolism changed their abundances, suggesting pivotal roles for growth under the respective conditions. -

Supplementary Information
Supplementary information (a) (b) Figure S1. Resistant (a) and sensitive (b) gene scores plotted against subsystems involved in cell regulation. The small circles represent the individual hits and the large circles represent the mean of each subsystem. Each individual score signifies the mean of 12 trials – three biological and four technical. The p-value was calculated as a two-tailed t-test and significance was determined using the Benjamini-Hochberg procedure; false discovery rate was selected to be 0.1. Plots constructed using Pathway Tools, Omics Dashboard. Figure S2. Connectivity map displaying the predicted functional associations between the silver-resistant gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S3. Connectivity map displaying the predicted functional associations between the silver-sensitive gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S4. Metabolic overview of the pathways in Escherichia coli. The pathways involved in silver-resistance are coloured according to respective normalized score. Each individual score represents the mean of 12 trials – three biological and four technical. Amino acid – upward pointing triangle, carbohydrate – square, proteins – diamond, purines – vertical ellipse, cofactor – downward pointing triangle, tRNA – tee, and other – circle. -

Supplementary Informations SI2. Supplementary Table 1
Supplementary Informations SI2. Supplementary Table 1. M9, soil, and rhizosphere media composition. LB in Compound Name Exchange Reaction LB in soil LBin M9 rhizosphere H2O EX_cpd00001_e0 -15 -15 -10 O2 EX_cpd00007_e0 -15 -15 -10 Phosphate EX_cpd00009_e0 -15 -15 -10 CO2 EX_cpd00011_e0 -15 -15 0 Ammonia EX_cpd00013_e0 -7.5 -7.5 -10 L-glutamate EX_cpd00023_e0 0 -0.0283302 0 D-glucose EX_cpd00027_e0 -0.61972444 -0.04098397 0 Mn2 EX_cpd00030_e0 -15 -15 -10 Glycine EX_cpd00033_e0 -0.0068175 -0.00693094 0 Zn2 EX_cpd00034_e0 -15 -15 -10 L-alanine EX_cpd00035_e0 -0.02780553 -0.00823049 0 Succinate EX_cpd00036_e0 -0.0056245 -0.12240603 0 L-lysine EX_cpd00039_e0 0 -10 0 L-aspartate EX_cpd00041_e0 0 -0.03205557 0 Sulfate EX_cpd00048_e0 -15 -15 -10 L-arginine EX_cpd00051_e0 -0.0068175 -0.00948672 0 L-serine EX_cpd00054_e0 0 -0.01004986 0 Cu2+ EX_cpd00058_e0 -15 -15 -10 Ca2+ EX_cpd00063_e0 -15 -100 -10 L-ornithine EX_cpd00064_e0 -0.0068175 -0.00831712 0 H+ EX_cpd00067_e0 -15 -15 -10 L-tyrosine EX_cpd00069_e0 -0.0068175 -0.00233919 0 Sucrose EX_cpd00076_e0 0 -0.02049199 0 L-cysteine EX_cpd00084_e0 -0.0068175 0 0 Cl- EX_cpd00099_e0 -15 -15 -10 Glycerol EX_cpd00100_e0 0 0 -10 Biotin EX_cpd00104_e0 -15 -15 0 D-ribose EX_cpd00105_e0 -0.01862144 0 0 L-leucine EX_cpd00107_e0 -0.03596182 -0.00303228 0 D-galactose EX_cpd00108_e0 -0.25290619 -0.18317325 0 L-histidine EX_cpd00119_e0 -0.0068175 -0.00506825 0 L-proline EX_cpd00129_e0 -0.01102953 0 0 L-malate EX_cpd00130_e0 -0.03649016 -0.79413596 0 D-mannose EX_cpd00138_e0 -0.2540567 -0.05436649 0 Co2 EX_cpd00149_e0 -

Supplementary Information
Supplementary Information Laboratory cultivation of acidophilic nanoorganisms. Physiological and bioinformatic dissection of a stable laboratory co-culture. Susanne Krause [1], Andreas Bremges [3,4], Philipp C. Münch [3,5], Alice C. McHardy [3] and Johannes Gescher* [1,2] [1] Department of Applied Biology, Karlsruhe Institute of Technology (KIT), Karlsruhe, Germany [2] Institute for Biological Interfaces, Karlsruhe Institute of Technology (KIT), Eggenstein- Leopoldshafen, Germany [3] Computational Biology of Infection Research, Helmholtz Centre for Infection Research, Braunschweig, Germany [4] German Center for Infection Research (DZIF), partner site Hannover-Braunschweig, Braunschweig, Germany [5] Max von Pettenkofer-Institute of Hygiene and Medical Microbiology, Ludwig-Maximilians- University of Munich, Munich, Germany SI Materials and Methods Quantification of cells using quantitative PCR The target sequences were amplified by PCR from genomic DNA of the enrichment cultures using primers see table S8. The amplified fragments contained overlapping regions as well as a BamHI site at the 5’ end and a SacI site at the 3’ end. Using the overlaps, the fragments were combined and cloned via the added restrictions sites into plasmid pAH95 1. Integration in the genome of E. coli DH5alphaZ1 was conducted as described before 1. For standard curve design, serial dilutions of E. coli DH5alphaZ1 cells containing the merged 23S target sequences of the ARMAN und Thermoplasmatales enrichments were prepared and cells were counted in a Neubauer counting chamber (Marienfeld, Lauda-Königshofen, Germany). DNA of the dilution series as well as of enrichment-cultures was extracted using the innuSPEED soil DNA kit (Analytic Jena, Jena, Germany) with minor modifications to the manufacturer´s instructions. -

SI Appendix Index 1
SI Appendix Index Calculating chemical attributes using EC-BLAST ................................................................................ 2 Chemical attributes in isomerase reactions ............................................................................................ 3 Bond changes …..................................................................................................................................... 3 Reaction centres …................................................................................................................................. 5 Substrates and products …..................................................................................................................... 6 Comparative analysis …........................................................................................................................ 7 Racemases and epimerases (EC 5.1) ….................................................................................................. 7 Intramolecular oxidoreductases (EC 5.3) …........................................................................................... 8 Intramolecular transferases (EC 5.4) ….................................................................................................. 9 Supporting references …....................................................................................................................... 10 Fig. S1. Overview …............................................................................................................................ -

Supporting Information for Proteomics DOI 10.1002/Pmic.200600987
Supporting Information for Proteomics DOI 10.1002/pmic.200600987 Lars Whlbrand, Birte Kallerhoff, Daniela Lange, Peter Hufnagel, Jrgen Thiermann, Richard Reinhardt and Ralf Rabus Functional proteomic view of metabolic regulation in “Aromatoleum aromaticum” strain EbN1 ª 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com Content 1. General proteome features Page Experimental outline of the presented study with “Aromatoleum aromaticum” 1-2 strain EbN1 (Fig. 1.1.) Spot pattern comparison of differently visualized 2DE gels (Fig. 1.2.) 1-3 Principle component analysis of the proteins for each analyzed 2D DIGE gel 1-4 of all main physiological groups (Fig. 1.3.) Fold changes in protein abundance of all identified proteins (Fig 1-5 Number of regulated as well as not regulated protein spots on the 2DE gels 1-6 and their share of total protein (Table 1.1.) Mean average ratio of not regulated protein spots of the three main 1-7 physiological groups (Table 1.2.) Mean average ratios of not regulated proteins, identified in all three main 1-8 physiological groups (Table 1.3.) Proteins identified in this study (Table 1.4.) 1-9 2. Cellular functions* 2.1. DNA replication, recombination and repair 2-2 2.1.1. DNA replication 2-2 2.2. Transcription 2-4 2.2.1. DNA-dependent RNA polymerase and sigma factors 2-5 2.2.2. Factors affecting RNA polymerase 2-5 2.2.3. Classes of transcriptional regulators 2-5 2.3. Translation 2-6 2.3.1. tRNA synthesis 2-6 2.3.2. tRNA/rRNA modification 2-7 2.3.3. -

Oup Proeng Gzy011 135..145 ++
Protein Engineering, Design & Selection, 2018, vol. 31 no. 4, pp. 135–145 doi: 10.1093/protein/gzy011 Advance Access Publication Date: 30 May 2018 Original Article Original Article A platform for chemical modification of mandelate racemase: characterization of the C92S/C264S and γ-thialysine 166 variants Downloaded from https://academic.oup.com/peds/article/31/4/135/5025590 by guest on 30 September 2021 Mitesh Nagar1, Himank Kumar1, and Stephen L. Bearne1,2,* 1Department of Biochemistry and Molecular Biology, Dalhousie University, Halifax, NS, Canada B3H 4R2, and 2Department of Chemistry, Dalhousie University, Halifax, NS, Canada B3H 4R2 *To whom correspondence should be addressed. E-mail: [email protected] Edited by Miroslaw Cygler Received 7 February 2018; Revised 28 April 2018; Editorial Decision 30 April 2018; Accepted 3 May 2018 Abstract Mandelate racemase (MR) serves as a paradigm for our understanding of enzyme-catalyzed deprotona- tion of a carbon acid substrate. To facilitate structure–function studies on MR using non-natural amino acid substitutions, we engineered the Cys92Ser/Cys264Ser variant (dmMR) as a platform for introdu- cingCysresiduesatspecific locations for subsequent covalent modification. While the highly reactive thiol of Cys furnishes a site for chemical modification, site-specificity requires that other Cys residues be non-reactive or replaced by a non-reactive amino acid, especially if chemical modification is con- ducted under denaturing conditions. The catalytic efficiency of dmMR is reduced only ~2-fold relative to wild-type MR, making dmMR a viable platform for the site-specificintroductionofCys.Asan example, the inactive Lys166Cys variant of dmMR was treated with ethylenimine under denaturing con- ditions to replace the Brønsted acid-base catalyst Lys 166 with the non-natural amino acid γ-thialysine. -

Impact of Temperature on the Time Required for the Establishment of Primordial Biochemistry, and for the Evolution of Enzymes
Impact of temperature on the time required for the establishment of primordial biochemistry, and for the evolution of enzymes Randy B. Stockbridge, Charles A. Lewis, Jr., Yang Yuan, and Richard Wolfenden1 Department of Biochemistry and Biophysics, University of North Carolina, Chapel Hill, NC 27599 Contributed by Richard Wolfenden, September 29, 2010 (sent for review August 1, 2010) All reactions are accelerated by an increase in temperature, but the magnitude of that effect on very slow reactions does not seem to have been fully appreciated. The hydrolysis of polysaccharides, for example, is accelerated 190,000-fold when the temperature is raised from 25 to 100 °C, while the rate of hydrolysis of phosphate monoester dianions increases 10,300,000-fold. Moreover, the slow- est reactions tend to be the most heat-sensitive. These tendencies collapse, by as many as five orders of magnitude, the time that would have been required for early chemical evolution in a warm environment. We propose, further, that if the catalytic effect of a “proto-enzyme”—like that of modern enzymes—were mainly enthalpic, then the resulting rate enhancement would have in- creased automatically as the environment became cooler. Several powerful nonenzymatic catalysts of very slow biological reactions, notably pyridoxal phosphate and the ceric ion, are shown to meet that criterion. Taken together, these findings greatly reduce the time that would have been required for early chemical evolution, countering the view that not enough time has passed for life to Fig. 1. Half-lives (t1∕2) and first order rate constants (k) of some biological have evolved to its present level of complexity. -

8-Barrel Enzymes John a Gerlt� and Frank M Raushely
252 Evolution of function in (b/a)8-barrel enzymes John A Gerltà and Frank M Raushely The (b/a)8-barrel is the most common fold in structurally closed, parallel b-sheet structure of the (b/a)8-barrel is characterized enzymes. Whether the functionally diverse formed from eight parallel (b/a)-units linked by hydrogen enzymes that share this fold are the products of either divergent bonds that form a cylindrical core. Despite its eightfold or convergent evolution (or both) is an unresolved question that pseudosymmetry, the packing within the interior of the will probably be answered as the sequence databases continue barrel is better described as four (b/a)2-subdomains in to expand. Recent work has examined natural, designed, and which distinct hydrophobic cores are located between the directed evolution of function in several superfamilies of (b/a)8- b-sheets and flanking a-helices [2]. barrel containing enzymes. The active sites are located at the C-terminal ends of the Addresses b-strands. So placed, the functional groups surround the ÃDepartments of Biochemistry and Chemistry, University of Illinois at active site and are structurally independent: the ‘old’ and Urbana-Champaign, 600 South Mathews Avenue, Urbana, ‘new’ enzymes retain functional groups at the ends of Illinois 61801, USA e-mail: [email protected] some b-strands, but others are altered to allow the ‘new’ yDepartment of Chemistry, Texas A&M University, PO Box 30012, activity [3]. With this blueprint, the (b/a)8-barrel is opti- College Station, Texas 77842-3012, USA mized for evolution of new functions. -
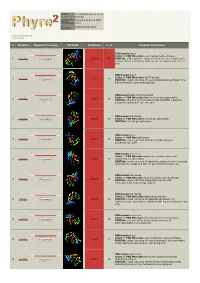
Phyre 2 Results for P29208
Email [email protected] Description P29208 Thu Jan 5 11:45:36 GMT Date 2012 Unique Job 0bd634b8f8620003 ID Detailed template information # Template Alignment Coverage 3D Model Confidence % i.d. Template Information PDB header:lyase Chain: A: PDB Molecule:o-succinylbenzoate synthase; 1 c3gc2A_ Alignment 100.0 86 PDBTitle: 1.85 angstrom crystal structure of o-succinylbenzoate synthase from2 salmonella typhimurium in complex with succinic acid PDB header:lyase Chain: A: PDB Molecule:tlr1174 protein; 2 c2oztA_ 100.0 20 Alignment PDBTitle: crystal structure of o-succinylbenzoate synthase from2 thermosynechococcus elongatus bp-1 PDB header:metal binding protein Chain: A: PDB Molecule:enzyme of enolase superfamily; 3 c3px5A_ 100.0 19 Alignment PDBTitle: structure of efi enolase target en500555, a putative dipeptide2 epimerase: apo structure PDB header:isomerase 4 c1jpmB_ Alignment 100.0 20 Chain: B: PDB Molecule:l-ala-d/l-glu epimerase; PDBTitle: l-ala-d/l-glu epimerase PDB header:lyase Chain: A: PDB Molecule:menc; 5 c2pgeA_ 100.0 23 Alignment PDBTitle: crystal structure of menc from desulfotalea psychrophila2 lsv54 PDB header:isomerase Chain: E: PDB Molecule:mandelate racemase/muconate 6 c3q45E_ Alignment 100.0 19 lactonizing enzyme family; PDBTitle: crystal structure of dipeptide epimerase from cytophaga hutchinsonii2 complexed with mg and dipeptide d-ala-l-val PDB header:isomerase Chain: A: PDB Molecule:chloromuconate cycloisomerase; 7 c1nu5A_ 100.0 16 Alignment PDBTitle: crystal structure of pseudomonas sp. p51 chloromuconate lactonizing2