Accepted Version
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Dr. Martin St. Maurice's Publications
Dr. Martin St. Maurice’s Publications 2013 Lin, Y., and St. Maurice, M. 2013. The structure of allophanate hydrolase from Granulibacter bethesdensis provides insights into substrate specificity in the amidase signature family. Biochemistry. 52: 690-700. 2012 Waldrop, G.L., Holden, H.M., and St. Maurice, M. 2012. The enzymes of biotin dependent CO2 metabolism: What structures reveal about their reaction mechanisms. Protein Science 21(11):1597-1619. Adina-Zada, A., Sereeruk, C., Jitrapakdee, S., Zeczycki, T.N., St. Maurice, M., Cleland, W.W., Wallace, J.C., and Attwood, P.V. 2012. Roles of Arg427 and Arg472 in the binding and allosteric effects of acetyl CoA in pyruvate carboxylase. Biochemistry 51(41): 1597-1619. 2011 Adina-Zada, A., Hazra, R., Sereeruk, C., Jitrapakdee, S., Zeczycki, T.N., St. Maurice, M., Cleland, W.W., Wallace, J.C., and Attwood, P.V. 2011. Probing the allosteric activation of pyruvate carboxylase using 2′,3′-O-(2,4,6-trinitrophenyl) adenosine 5′-triphosphate as a fluorescent mimic of the allosteric activator acetyl CoA. Arch. Biochem. Biophys. 117-126. Zeczycki, T.N., Menefee, A.L., Jitrapakdee, S., Wallace, J.C., Attwood, P.V., St. Maurice, M. and Cleland, W.W. 2011. Activation and inhibition of pyruvate carboxylase from Rhizobium etli. Biochemistry. 9694-9707. Lietzan, A.D., Menefee, A.L., Zeczycki, T.N., Kumar, S., Attwood, P.V., Wallace, J.C., Cleland, W.W. and St. Maurice, M. 2011. Interaction between the biotin carrier domain and the biotin carboxylase domain in the structure of Rhizobium etli pyruvate carboxylase. Biochemistry. 9708-9723. Zeczycki, T.N., Menefee, A.L., Adina-Zada, A., Surinya, K.H., Wallace, J.C., Attwood, P.V., St. -

SI Appendix Index 1
SI Appendix Index Calculating chemical attributes using EC-BLAST ................................................................................ 2 Chemical attributes in isomerase reactions ............................................................................................ 3 Bond changes …..................................................................................................................................... 3 Reaction centres …................................................................................................................................. 5 Substrates and products …..................................................................................................................... 6 Comparative analysis …........................................................................................................................ 7 Racemases and epimerases (EC 5.1) ….................................................................................................. 7 Intramolecular oxidoreductases (EC 5.3) …........................................................................................... 8 Intramolecular transferases (EC 5.4) ….................................................................................................. 9 Supporting references …....................................................................................................................... 10 Fig. S1. Overview …............................................................................................................................ -

Supporting Information for Proteomics DOI 10.1002/Pmic.200600987
Supporting Information for Proteomics DOI 10.1002/pmic.200600987 Lars Whlbrand, Birte Kallerhoff, Daniela Lange, Peter Hufnagel, Jrgen Thiermann, Richard Reinhardt and Ralf Rabus Functional proteomic view of metabolic regulation in “Aromatoleum aromaticum” strain EbN1 ª 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com Content 1. General proteome features Page Experimental outline of the presented study with “Aromatoleum aromaticum” 1-2 strain EbN1 (Fig. 1.1.) Spot pattern comparison of differently visualized 2DE gels (Fig. 1.2.) 1-3 Principle component analysis of the proteins for each analyzed 2D DIGE gel 1-4 of all main physiological groups (Fig. 1.3.) Fold changes in protein abundance of all identified proteins (Fig 1-5 Number of regulated as well as not regulated protein spots on the 2DE gels 1-6 and their share of total protein (Table 1.1.) Mean average ratio of not regulated protein spots of the three main 1-7 physiological groups (Table 1.2.) Mean average ratios of not regulated proteins, identified in all three main 1-8 physiological groups (Table 1.3.) Proteins identified in this study (Table 1.4.) 1-9 2. Cellular functions* 2.1. DNA replication, recombination and repair 2-2 2.1.1. DNA replication 2-2 2.2. Transcription 2-4 2.2.1. DNA-dependent RNA polymerase and sigma factors 2-5 2.2.2. Factors affecting RNA polymerase 2-5 2.2.3. Classes of transcriptional regulators 2-5 2.3. Translation 2-6 2.3.1. tRNA synthesis 2-6 2.3.2. tRNA/rRNA modification 2-7 2.3.3. -

Oup Proeng Gzy011 135..145 ++
Protein Engineering, Design & Selection, 2018, vol. 31 no. 4, pp. 135–145 doi: 10.1093/protein/gzy011 Advance Access Publication Date: 30 May 2018 Original Article Original Article A platform for chemical modification of mandelate racemase: characterization of the C92S/C264S and γ-thialysine 166 variants Downloaded from https://academic.oup.com/peds/article/31/4/135/5025590 by guest on 30 September 2021 Mitesh Nagar1, Himank Kumar1, and Stephen L. Bearne1,2,* 1Department of Biochemistry and Molecular Biology, Dalhousie University, Halifax, NS, Canada B3H 4R2, and 2Department of Chemistry, Dalhousie University, Halifax, NS, Canada B3H 4R2 *To whom correspondence should be addressed. E-mail: [email protected] Edited by Miroslaw Cygler Received 7 February 2018; Revised 28 April 2018; Editorial Decision 30 April 2018; Accepted 3 May 2018 Abstract Mandelate racemase (MR) serves as a paradigm for our understanding of enzyme-catalyzed deprotona- tion of a carbon acid substrate. To facilitate structure–function studies on MR using non-natural amino acid substitutions, we engineered the Cys92Ser/Cys264Ser variant (dmMR) as a platform for introdu- cingCysresiduesatspecific locations for subsequent covalent modification. While the highly reactive thiol of Cys furnishes a site for chemical modification, site-specificity requires that other Cys residues be non-reactive or replaced by a non-reactive amino acid, especially if chemical modification is con- ducted under denaturing conditions. The catalytic efficiency of dmMR is reduced only ~2-fold relative to wild-type MR, making dmMR a viable platform for the site-specificintroductionofCys.Asan example, the inactive Lys166Cys variant of dmMR was treated with ethylenimine under denaturing con- ditions to replace the Brønsted acid-base catalyst Lys 166 with the non-natural amino acid γ-thialysine. -

Impact of Temperature on the Time Required for the Establishment of Primordial Biochemistry, and for the Evolution of Enzymes
Impact of temperature on the time required for the establishment of primordial biochemistry, and for the evolution of enzymes Randy B. Stockbridge, Charles A. Lewis, Jr., Yang Yuan, and Richard Wolfenden1 Department of Biochemistry and Biophysics, University of North Carolina, Chapel Hill, NC 27599 Contributed by Richard Wolfenden, September 29, 2010 (sent for review August 1, 2010) All reactions are accelerated by an increase in temperature, but the magnitude of that effect on very slow reactions does not seem to have been fully appreciated. The hydrolysis of polysaccharides, for example, is accelerated 190,000-fold when the temperature is raised from 25 to 100 °C, while the rate of hydrolysis of phosphate monoester dianions increases 10,300,000-fold. Moreover, the slow- est reactions tend to be the most heat-sensitive. These tendencies collapse, by as many as five orders of magnitude, the time that would have been required for early chemical evolution in a warm environment. We propose, further, that if the catalytic effect of a “proto-enzyme”—like that of modern enzymes—were mainly enthalpic, then the resulting rate enhancement would have in- creased automatically as the environment became cooler. Several powerful nonenzymatic catalysts of very slow biological reactions, notably pyridoxal phosphate and the ceric ion, are shown to meet that criterion. Taken together, these findings greatly reduce the time that would have been required for early chemical evolution, countering the view that not enough time has passed for life to Fig. 1. Half-lives (t1∕2) and first order rate constants (k) of some biological have evolved to its present level of complexity. -
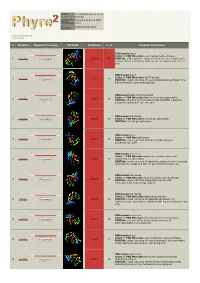
Phyre 2 Results for P29208
Email [email protected] Description P29208 Thu Jan 5 11:45:36 GMT Date 2012 Unique Job 0bd634b8f8620003 ID Detailed template information # Template Alignment Coverage 3D Model Confidence % i.d. Template Information PDB header:lyase Chain: A: PDB Molecule:o-succinylbenzoate synthase; 1 c3gc2A_ Alignment 100.0 86 PDBTitle: 1.85 angstrom crystal structure of o-succinylbenzoate synthase from2 salmonella typhimurium in complex with succinic acid PDB header:lyase Chain: A: PDB Molecule:tlr1174 protein; 2 c2oztA_ 100.0 20 Alignment PDBTitle: crystal structure of o-succinylbenzoate synthase from2 thermosynechococcus elongatus bp-1 PDB header:metal binding protein Chain: A: PDB Molecule:enzyme of enolase superfamily; 3 c3px5A_ 100.0 19 Alignment PDBTitle: structure of efi enolase target en500555, a putative dipeptide2 epimerase: apo structure PDB header:isomerase 4 c1jpmB_ Alignment 100.0 20 Chain: B: PDB Molecule:l-ala-d/l-glu epimerase; PDBTitle: l-ala-d/l-glu epimerase PDB header:lyase Chain: A: PDB Molecule:menc; 5 c2pgeA_ 100.0 23 Alignment PDBTitle: crystal structure of menc from desulfotalea psychrophila2 lsv54 PDB header:isomerase Chain: E: PDB Molecule:mandelate racemase/muconate 6 c3q45E_ Alignment 100.0 19 lactonizing enzyme family; PDBTitle: crystal structure of dipeptide epimerase from cytophaga hutchinsonii2 complexed with mg and dipeptide d-ala-l-val PDB header:isomerase Chain: A: PDB Molecule:chloromuconate cycloisomerase; 7 c1nu5A_ 100.0 16 Alignment PDBTitle: crystal structure of pseudomonas sp. p51 chloromuconate lactonizing2 -

The Enolase Superfamily: a General Strategy for Enzyme-Catalyzed Abstraction of the R-Protons of Carboxylic Acids†
+ + Biochemistry 1996, 35, 16489-16501 16489 The Enolase Superfamily: A General Strategy for Enzyme-Catalyzed Abstraction of the R-Protons of Carboxylic Acids† ,‡ §,| ⊥,# Patricia C. Babbitt,* Miriam S. Hasson, Joseph E. Wedekind, David R. J. Palmer,r William C. Barrett,r ⊥ ⊥ § ‡ , George H. Reed, Ivan Rayment, Dagmar Ringe, George L. Kenyon, and John A. Gerlt* r Department of Pharmaceutical Chemistry, UniVersity of California, San Francisco, California 94143-0446, Departments of Biochemistry and Chemistry and Rosenstiel Center for Basic Biomedical Research, Brandeis UniVersity, Waltham, Massachusetts 02154-9110, The Institute for Enzyme Research and Department of Biochemistry, UniVersity of Wisconsin, Madison, Wisconsin 53706, and Department of Biochemistry, UniVersity of Illinois, Urbana, Illinois 61801 ReceiVed July 5, 1996X ABSTRACT: We have discovered a superfamily of enzymes related by their ability to catalyze the abstraction of the R-proton of a carboxylic acid to form an enolic intermediate. Although each reaction catalyzed by these enzymes is initiated by this common step, their overall reactions (including racemization, â-elimination of water, â-elimination of ammonia, and cycloisomerization) as well as the stereochemical consequences (syn Vs anti) of the â-elimination reactions are diverse. Analysis of sequence and structural similarities among these proteins suggests that all of their chemical reactions are mediated by a common active site architecture modified through evolution to allow the enolic intermediates to partition to different products in their respective active sites Via different overall mechanisms. All of these enzymes retain the ability to catalyze the thermodynamically difficult step of proton abstraction. These homologous proteins, designated the “enolase superfamily”, include enolase as well as more metabolically specialized enzymes: mandelate racemase, galactonate dehydratase, glucarate dehydratase, muconate-lactonizing enzymes, N-acylamino acid racemase, â-methylaspartate ammonia-lyase, and o-succinylbenzoate synthase. -

Α-Methylacyl-Coa Racemase and Argininosuccinate Lyase
A460etukansi.fm Page 1 Friday, May 26, 2006 11:40 AM A 460 OULU 2006 A 460 UNIVERSITY OF OULU P.O. Box 7500 FI-90014 UNIVERSITY OF OULU FINLAND ACTA UNIVERSITATIS OULUENSIS ACTA UNIVERSITATIS OULUENSIS ACTA A SERIES EDITORS SCIENTIAE RERUM Prasenjit Bhaumik NATURALIUM Prasenjit Bhaumik Prasenjit ASCIENTIAE RERUM NATURALIUM Professor Mikko Siponen PROTEIN CRYSTALLOGRAPHIC BHUMANIORA STUDIES TO UNDERSTAND Professor Harri Mantila THE REACTION MECHANISM CTECHNICA Professor Juha Kostamovaara OF ENZYMES: DMEDICA α Professor Olli Vuolteenaho -METHYLACYL-CoA RACEMASE AND ESCIENTIAE RERUM SOCIALIUM Senior assistant Timo Latomaa ARGININOSUCCINATE LYASE FSCRIPTA ACADEMICA Communications Officer Elna Stjerna GOECONOMICA Senior Lecturer Seppo Eriksson EDITOR IN CHIEF Professor Olli Vuolteenaho EDITORIAL SECRETARY Publication Editor Kirsti Nurkkala FACULTY OF SCIENCE, DEPARTMENT OF BIOCHEMISTRY, BIOCENTER OULU, ISBN 951-42-8090-3 (Paperback) UNIVERSITY OF OULU ISBN 951-42-8091-1 (PDF) ISSN 0355-3191 (Print) ISSN 1796-220X (Online) ACTA UNIVERSITATIS OULUENSIS A Scientiae Rerum Naturalium 460 PRASENJIT BHAUMIK PROTEIN CRYSTALLOGRAPHIC STUDIES TO UNDERSTAND THE REACTION MECHANISM OF ENZYMES: α-METHYLACYL-COA RACEMASE AND ARGININOSUCCINATE LYASE Academic Dissertation to be presented with the assent of the Faculty of Science, University of Oulu, for public discussion in Kuusamonsali (Auditorium YB210), Linnanmaa, on June 6th, 2006, at 12 noon OULUN YLIOPISTO, OULU 2006 Copyright © 2006 Acta Univ. Oul. A 460, 2006 Supervised by Professor Rik Wierenga Reviewed -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S. -

Assay of the Prostate Cancer Biomarker A-Methylacyl Coenzyme a Racemase (AMACR)
Assay of the Prostate Cancer Biomarker a-Methylacyl Coenzyme A Racemase (AMACR) by Dahmane Ouazia Submitted in partial fulfillment of the requirements for the degree of Master of Science at Dalhousie University Halifax, Nova Scotia June 2008 © Copyright by Dahmane Ouazia, 2008 Library and Bibliotheque et 1*1 Archives Canada Archives Canada Published Heritage Direction du Branch Patrimoine de I'edition 395 Wellington Street 395, rue Wellington Ottawa ON K1A0N4 Ottawa ON K1A0N4 Canada Canada Your file Votre reference ISBN: 978-0-494-43994-4 Our file Notre reference ISBN: 978-0-494-43994-4 NOTICE: AVIS: The author has granted a non L'auteur a accorde une licence non exclusive exclusive license allowing Library permettant a la Bibliotheque et Archives and Archives Canada to reproduce, Canada de reproduire, publier, archiver, publish, archive, preserve, conserve, sauvegarder, conserver, transmettre au public communicate to the public by par telecommunication ou par Plntemet, prefer, telecommunication or on the Internet, distribuer et vendre des theses partout dans loan, distribute and sell theses le monde, a des fins commerciales ou autres, worldwide, for commercial or non sur support microforme, papier, electronique commercial purposes, in microform, et/ou autres formats. paper, electronic and/or any other formats. The author retains copyright L'auteur conserve la propriete du droit d'auteur ownership and moral rights in et des droits moraux qui protege cette these. this thesis. Neither the thesis Ni la these ni des extraits substantiels de nor substantial extracts from it celle-ci ne doivent etre imprimes ou autrement may be printed or otherwise reproduits sans son autorisation. -

Discovery of High Affinity Receptors for Dityrosine Through Inverse Virtual Screening and Docking and Molecular Dynamics
Article Discovery of High Affinity Receptors for Dityrosine through Inverse Virtual Screening and Docking and Molecular Dynamics Fangfang Wang 1,*,†, Wei Yang 2,3,† and Xiaojun Hu 1,* 1 School of Life Science, Linyi University, Linyi 276000, China; [email protected] 2 Department of Microbiology, Biomedicine Discovery Institute, Monash University, Clayton, VIC 3800, Australia, [email protected] 3 Arieh Warshel Institute of Computational Biology, the Chinese University of Hong Kong, 2001 Longxiang Road, Longgang District, Shenzhen 518000, China * Corresponding author: [email protected] † These authors contributed equally to this work. Received: 09 December 2018; Accepted: 23 December 2018; Published: date Table S1. Docking affinity scores for cis-dityrosine binding to binding proteins. Target name PDB/UniProtKB Type Affinity (kcal/mol) Galectin-1 1A78/P56217 Lectin -6.2±0.0 Annexin III 1AXN/P12429 Calcium/phospholipid Binding Protein -7.5±0.0 Calmodulin 1CTR/P62158 Calcium Binding Protein -5.8±0.0 Seminal Plasma Protein Pdc-109 1H8P/P02784 Phosphorylcholine Binding Protein -6.6±0.0 Annexin V 1HAK/P08758 Calcium/phospholipid Binding -7.4±0.0 Alpha 1 antitrypsin 1HP7/P01009 Protein Binding -7.6±0.0 Histidine-Binding Protein 1HSL/P0AEU0 Binding Protein -6.3±0.0 Intestinal Fatty Acid Binding Protein 1ICN/P02693 Binding Protein(fatty Acid) -9.1±0.0* Migration Inhibitory Factor-Related Protein 14 1IRJ/P06702 Metal Binding Protein -7.0±0.0 Lysine-, Arginine-, Ornithine-Binding Protein 1LST/P02911 Amino Acid Binding Protein -6.5±0.0 -

The Chemistry and Evolution of Enzyme Function
The Chemistry and Evolution of Enzyme Function: Isomerases as a Case Study Sergio Mart´ınez Cuesta EMBL - European Bioinformatics Institute Gonville and Caius College University of Cambridge A thesis submitted for the degree of Doctor of Philosophy 31st July 2014 This dissertation is the result of my own work and contains nothing which is the outcome of work done in collaboration except where specifically indicated in the text. No part of this dissertation has been submitted or is currently being submitted for any other degree or diploma or other qualification. This thesis does not exceed the specified length limit of 60.000 words as defined by the Biology Degree Committee. This thesis has been typeset in 12pt font using LATEX according to the specifications de- fined by the Board of Graduate Studies and the Biology Degree Committee. Cambridge, 31st July 2014 Sergio Mart´ınezCuesta To my parents and my sister Contents Abstract ix Acknowledgements xi List of Figures xiii List of Tables xv List of Publications xvi 1 Introduction 1 1.1 Chemistry of enzymes . .2 1.1.1 Catalytic sites, mechanisms and cofactors . .3 1.1.2 Enzyme classification . .5 1.2 Evolution of enzyme function . .6 1.3 Similarity between enzymes . .8 1.3.1 Comparing sequences and structures . .8 1.3.2 Comparing genomic context . .9 1.3.3 Comparing biochemical reactions and mechanisms . 10 1.4 Isomerases . 12 1.4.1 Metabolism . 13 1.4.2 Genome . 14 1.4.3 EC classification . 15 1.4.4 Applications . 18 1.5 Structure of the thesis . 20 2 Data Resources and Methods 21 2.1 Introduction .