Investigation of Lyophilized Antibody Formulations to Enable Short Freeze-Drying Cycles and Storage at Room Temperature
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Thermal Behavior Characterization of a Sugar-Based Model System and Commercial Confections Across the Stages of Sugar Cooking
THERMAL BEHAVIOR CHARACTERIZATION OF A SUGAR-BASED MODEL SYSTEM AND COMMERCIAL CONFECTIONS ACROSS THE STAGES OF SUGAR COOKING BY MELISSA WANG THESIS Submitted in partial fulfillment of the requirements for the degree of Master of Science in Food Science and Human Nutrition with a concentration in Food Science in the Graduate College of the University of Illinois at Urbana-Champaign, 2017 Urbana, Illinois Adviser: Professor Shelly J. Schmidt Abstract The stages of sugar cooking, although long-existing and widespread in the confection industry, are lacking in thermal behavior profile descriptions, which are crucial to confection functionality. Thermal behavior parameters, such as the glass transition temperature (Tg), are indicative of confection material structure and textural behavior. Tg plays an important role in governing the quality and shelf life of sugar-based confection, and is influenced by moisture content, formulation, and other factors. This study aimed to connect thermal behavior parameters to the stages of sugar cooking. Thus, the objective of this research was to investigate the thermal behavior of the six stages of sugar cooking, as well as representative commercial confections from each stage. A model sugar-based confectionery system was developed and representative commercial confections belonging to each stage of sugar cooking were selected. The model system consisted of a 70:30 ratio of sucrose to corn syrup and a 70:30 ratio of solids to moisture. To investigate the thermal behavior of the stages of sugar cooking, differential scanning calorimetry (DSC), moisture content, and water activity analyses were conducted for the model system and representative commercial confections. The average Tg midpoint of the model system increased from thread to hard crack stage, corresponding to loss of water from increased cooking time and temperature. -

Caramelization Processes in Sugar Glasses and Sugar Polycrystals
New Physics: Sae Mulli (The Korean Physical Society), DOI: 10.3938/NPSM.62.761 Volume 62, Number 7, 2012¸ 7Z4, pp. 761∼767 Caramelization Processes in Sugar Glasses and Sugar Polycrystals Jeong-Ah Seo · Hyun-Joung Kwon · Dong-Myeong Shin · Hyung Kook Kim · Yoon-Hwae Hwang∗ Department of Nanomaterials Engineering & BK21 Nano Fusion Technology Division, Pusan National University, Miryang 627-706 (Received 3 April 2012 : revised 3 June 2012 : accepted 2 July 2012) We studied the chemical dehydration processes due to the caramelization in sugar glasses and sugar polycrystals. The dehydration processes of three monosaccharide sugars (fructose, galactose, and glucose) and three disaccharide sugars (sucrose, maltose, and trehalose) were compared by using a thermogravimetic-differential thermal analyzer to measure the mass reduction. The amounts of mass reductions in sugar glasses were larger than those in sugar polycrystals. However, the amount of mass reduction in trehalose glasses was smaller than that in trehalose polycrystals. This unique dehydration property of trehalose glasses may be related to the high glass transition temperature, which might be related to a superior bioprotection ability of trehalose. PACS numbers: 87.14.Df, 81.70.Pg, 64.70.P Keywords: Dehydration, Caramelization, Sugar glass, Trehalose, Disaccharide I. INTRODUCTION higher than that of other disaccharides [6,7]. Therefore, the viscosity of trehalose is higher than that of other There are many strategies in nature for the long-term sugars at a given temperature. Several researchers have survival and storage of organisms, and a bio-protection pointed out that the high glass-transition temperature of effect is one of the most interesting among those long- trehalose may contribute to the preservation of biologi- term survival and storage mechanisms. -

Making Sugar Into Glass
Glass KS2: Making sugar into glass Lesson Objective: To make some sugar glass and learn about the properties of glass. Science National Curriculum links: Y3 Science: Light – recognise that they need light to see things and that dark is the absence of light; notice that light is reflected from surfaces. Y4 Science: States of matter – compare and group materials together, according to whether they are solids, liquids or gases; observe that some materials change state when they are heated or cooled. Y5 Science: Properties and changes of materials – Give reasons, based on comparative and fair tests, for the particular uses of everyday materials. Y6 Science: Light – recognise that light travels in straight lines. Resources: • 790g granulated sugar • 475ml water • 240ml light corn syrup • ¼ tsp cream of tartar • Saucepan • Hob or camping stove • Silicone spatula • Confectionery or jam thermometer • Baking sheet • Aluminium foil or cooking spray Time required: 60 mins plus 60 mins cooling NOTE: this activity heats sugar to high temperatures so ensure proper adult supervision is available. You may want to carry out this activity as a class demonstration. Introduction to Activity: One of the wonderful properties of glass is that it is brittle and so breaks into lots of little shards. These shards are sharp and dangerous, so alternatives had to be found for doing stunts in films. The answer is to use sugar! Breakaway or sugar glass can be formed into many different shapes, looks and sounds like glass and when it breaks it does not cause injury – ideal for action films! In this activity your class will make some sugar glass of your own! Main Activity: Firstly, prepare a large baking sheet, line the bottom with a spray of cooking oil or a piece of aluminium foil and put to one side. -

Shakes Cocktails Hot Drinks Beers Soft Drinks Wine
LONG ISLAND ICE TEA 6.00 Five white spirits, lemon juice & cola WINE COCKTAILS TEXAN CREAM SODA 5.30 FIREBALLER 5.95 Titos Handcrafted Texan vodka with WHITE 175ml 250ml Bottle Fireball cinnamon whiskey with lime juice and Soda Folk Cream Soda SHORT MILE BAY SAUVIGNON Australia 4.95 6.35 17.95 cloudy apple and rhubarb CHERRY COLA 5.30 A succulent Sauv with a crisp citrus finish Old J Cherry, fresh lime & cola RHUBARB & HIBISCUS HIBALL 5.95 KLEINE ZALZE CHENIN BLANC South Africa 5.20 6.90 19.50 RUM PUNCH 6.95 Titos Handcrafted Texan vodka with One of the best examples of a Chenin we could get our hands on. Pusser, Morgan’s Spiced Rum, Amaretto, Rhubarb & Hibiscus tonic Baked apple balanced with citrus and honey melon pineapple juice, orange juice, lime & ginger QUINDI PINOT GRIGIO Italy 5.45 7.35 21.65 syrup NEGRONI 5.95 This wine is way too drinkable! KIR ROYALE 6.25 Soft, crisp and a finish that leaves you wanting more Campari, Martini Rosso, Beefeater Gin Prosecco & Chambord VAVASOUR SAUVIGNON BLANC New Zealand 5.95 7.75 23.45 ESPRESSO MARTINI 6.25 What New Zealand is famous for: heaps of FRENCH 75 6.55 character with tropical fruit on the finish Absolut Vanilla vodka shaken with Beefeater gin, lemon juice, sweetened cold brew coffee Prosecco & a dash of sugar ERRAZURIZ CHARDONNAY Chile 6.25 8.45 25.95 Rich, buttery and soft as a tender embrace AMARETTO SOUR 5.95 RHUBARB GIN FIZZ 5.95 Amaretto, lemon juice, egg white and sugar Beefeater Gin, rhubarb purée, lemon juice, egg white & sugar RED 175ml 250ml Bottle MOJITO 6.10 GODFATHER SOUR -

Happy Hour & Mimosas $5 Every Day 7Am - Noon $5
Select Cocktails, Happy Select Cocktails, Martinis, Margaritas, Martinis, Margaritas, Wines, & Frozen Drinks Hour Wines, & Frozen Drinks $6 Monday - Friday • 3-7 pm $6 Beer Craft Beers $5.25 Resurrection (12oz CAN, Brewer’s Art Balto.) Loose Cannon (IPA, Heavy Seas) Doggie Style (APA, Flying Dog Brewery) Samuel Adams (Lager, Boston Beer Co) Angry Orchard (Cider, Boston Beer Co) Domestic Premium Imports $4.25 $5.25 Natty Boh (16oz Can) - Bud Light - Miller Lite Peroni - Stella Artois - Blue Moon Yuengling - Odoul’s (non-alcoholic) Guinness Stout - Corona Extra - Heineken Wines Dry White Glass Bottle Dry Red Chardonnay, CA’DONINI (Italy) 8.00 28.00 Cabernet Sauvignon, FOLONARI (Italy) 9.50 35.00 A great seafood wine. Pairs well with grilled fish, crab cakes, Well matched with red meats, especially char-grilled steak and smoked foods poultry and creamy pasta dishes. Cabernet Sauvignon, KUNDE (Sonoma Valley) 11.50 44.00 Chardonnay, JOSH CELLARS (California) 9.00 32.00 Bouquet of black currant, cinnamon and berries. Good balance, uplifting acidity & a low finish of pear & creme brulée Merlot, Josh Cellars (California) 9.50 35.00 Pinot Grigio SANTA MARGHERITA (Italy) 12.50 48.00 A pop of ripe plum and berry flavors yet mellow. Great with a pink and juicy Ideal with seafood or shellfish salads, pasta and rice dishes char-grilled steak. Bouquet ripe plum, cassis, sage & subtle cedar Pinot Grigio, Spasso (Italy) 8.00 28.00 Merlot, FOLONARI (Italy) 9.00 32.00 Great alongside seafood, light pastas and pork dishes Bright red with berry driven character, a little spicy and hints of vanilla Semi-Sweet Shiraz, ROSEMOUNT ESTATES (S. -

Holiday Inspiration 2017
Holiday Inspiration 2017 www.footerscatering.com 303.762.1410 [email protected] Be Inspired We hope our seasonal menus inspire Love What You Do your perfect holiday celebration! With your vision in mind, our full service team will take care of the rest: That is our mission at Footers Catering. We are committed to creating a great place • Customized menu with your favorite items by our Culinary Crusaders to work because we believe that when our • Exceptional service from set-up to clean-up by our Hospitality Heroes Perfect seasonal cocktails from our Mix Masters team loves what they do, they are just as • • On-site cooking by our talented Chefs de Par-Tay excited about our events as you are. • Timeline, diagram and vendor planning with our Detailed Divas • Linen, rental and decor coordination by our Production Team From customizing your perfect menu to Every event is unique and we specialize in designing proposals around handling special requests with a smile, our your specific needs. For general planning purposes, all-inclusive pricing team will work hard and have fun creating an with complete menu, hospitality team, bar service (alcohol not included), chefs to cook all menu items fresh on-site, rentals, linen and unforgettable event for you and your guests! applicable tax and surcharge, is estimated in the following ranges: • Cocktail Receptions - $50-$70 per guest • Buffet or Station Receptions - $60-$80 per guest • Plated or Family-Style Receptions - $70-$90 per guest Our Event Coordinators would love to discuss how we can help -
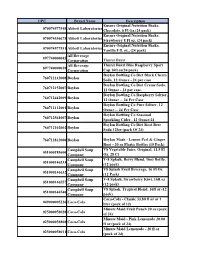
UPC Brand Name Description 070074577548
UPC Brand Name Description Ensure Original Nutrition Shake, 070074577548 Abbott Laboratories Chocolate, 8 Fl Oz (24 pack) Ensure Original Nutrition Shake, 070074536675 Abbott Laboratories Strawberry 8 Fl oz, (24 pack) Ensure Original Nutrition Shake, 070074577531 Abbott Laboratories Vanilla 8 fl. oz., (24 pack) All Beverage 897740000045 Corporation Thurst Burst All Beverage Thurst Burst Blue Raspberry Sport 897740000038 Corporation Cap 16fl oz(24 pack) Boylan Bottling Co Diet Black Cherry 760712142008 Boylan Soda, 12 Ounce - 24 per case Boylan Bottling Co Diet Creme Soda, 760712152007 Boylan 12 Ounce - 24 per case. Boylan Bottling Co Raspberry Seltzer, 760712442009 Boylan 12 Ounce -- 24 Per Case Boylan Bottling Co Pure Seltzer, 12 760712112001 Boylan Ounce -- 24 Per Case Boylan Bottling Co Seasonal 760712561007 Boylan Sparkling Cider , 12 Ounce-24 Boylan Bottling Co Diet Root Beer 760712102002 Boylan Soda 12foz (pack Of 24) 760712812000 Boylan Boylan Mash - Lemon Peel & Ginger Root - 20 oz Plastic Bottles (15 Pack) Campbell Soup V8 Vegetable Juice, Original, 11.5 Fl 051000185600 Company Oz, 28 Ct Campbell Soup V-8 Splash, Berry Blend, 16oz Bottle, 051000146533 Company (12 pack) Campbell Soup V8 Splash Fruit Beverage, 16 Fl Oz 051000146632 Company (12 Pack) Campbell Soup V-8 Splash, Strawberry Kiwi, 16fl oz 051000146557 Company (12 pack) Campbell Soup V8 Splash, Tropical Blend, 16fl oz (12 051000146540 Company pack) Coca-Cola - Classic 33.80 fl oz or 1 049000005226 Coca-Cola liter (pack of 12) Minute Maid Fruit Punch 20 oz (pack 025000058028 Coca-Cola of 24) Minute Maid - Pink Lemonade 20.00 025000058868 Coca-Cola fl oz (pack of 24) Minute Maid Lemonade - 20 fl oz 025000058011 Coca-Cola (pack of 24) Coca Cola, Cherry Coke Zero 20.0 049000047547 Coca-Cola Oz. -

Discovering Science and Technology of Glass from Candy Making
Session 3264 Discovering the Science and Technology of Glass Formation from Candy Making H. Jain and I. H. Jain Department of Materials Science and Engineering Lehigh University, Bethlehem, PA 18015 Abstract Glass is a material that has been used for centuries in numerous common, consumer applications. In recent decades, it has become one of the key high-tech materials for applications in thermal insulation, optical communication and microelectronics. Notwithstanding its importance, the average student's understanding of glass science and technology remains very primitive, partly because of its ill-defined structure and complex behavior. Therefore, to illustrate the underlying principles of this class of materials, we have proposed a set of laboratory demonstrations involving sucrose, water and corn starch (the major ingredients of common candy). These low cost simple experiments provide a convenient paradigm for the numerous features of a prototypic glass. The students' feedback has been very positive as they find the unusual experiments exciting, and can relate to the results from previous experiences. Introduction Glass is a common material that has been known to man for several thousand years. Early applications of glass such as in windows, containers, lenses, tableware, jewelry depended on their transparency, luster and durability. Many recent high-tech applications such as optical fiber for high-speed communication or dielectrics in microelectronics also exploit some such qualities, but these are neither necessary nor sufficient attributes for a material to be called as glass. A general definition of glass is that it is 'a solid with liquid like structure', 'a noncrystalline solid' or simply 'an amorphous solid'.1 None of these definitions is very helpful to typical engineering students, even the ones majoring in Materials Science and Engineering, so that they can appreciate the characteristics of this important class of material. -

Candy Glass Recipe, Instructions and Worksheet
Candy Glass Making Demonstration for Classroom or Science Activity. Prepared by Dr. William Heffner, International Materials Institute for Glass (IMI-NFG), Lehigh University. Glass Education Webpage: www.lehigh.edu/imi/libraryglasseducation.html ____________________________________________ Candy Glass Recipe: Making sugar glass is a very simple. It is essentially a mixture of sugar(s) and water heated to the right temperature to first dissolve the sugar and then boil off most of the water, stopping at the appropriate temperature to form a “glass” on cooling. The temperature at which we stop boiling the mixture is directly linked to the residual water content which must be low enough to form the glass state of a hard candy. If the temperature is too low a softer, non glass, taffy-like candy will result instead (or even just a syrup). Some care must be taken to avoid having the highly concentrated sugar solution form sugar crystals on cooling rather than remain in the “frozen liquid” state of the glass. Stirring can initiate this crystallization in high sucrose recipes. We will discuss crystallization in more detail later. For now let us begin with the simple recipe for hard candy below. It uses a mixture of sugars so crystallization is less likely to occur and should not pose any problem if you follow the recipe and avoid excess stirring. Some of the ingredients and equipment you will need for the making of candy glass. Recipe: approx. Weight US volume (metric grams) measure Cane sugar 120 g ½ cup (sucrose) Karo corn 60 g ~ ¼ cup syrup 4 teaspoons Water 20 g (2/3 oz) In Preparation: In addition to the ingredients listed above you will want to have the following equipment available: candy thermometer (digital recommended) 1 quart sauce pan (or 400 ml Pyrex beaker) spoon dish of cold water for testing the “doneness” (sort ball / hard ball) notepad and pen to record temperature and test data Candy Glass Making Demonstration for Classroom or Science Activity. -

Sugar Confectionary
Candy Mystical Consumers attach a certain sense of magic and wonder to candies. This isn't surprising, considering all of the many unique varieties of candy started out as a concentrated solution of sugar in water. Sugar Confectioners need an understanding of basic confectionery structure and technique to transform these simple ingredients into confectionery delights. Essential Ingredients The primary ingredient in candy is sugar as sucrose. Turning the sugar into candy involves dissolving it in water, concentrating this solution through cooking, and subsequently allowing the mass either to form mutable solid or to recrystallize. Key elements in the candy making process are in sugar's physical properties. Specifically its solubility with its effect on the water's boiling point. At room temperature, about 2 kilos of sugar will dissolve in 1 kilo of water. At higher temperatures, more sugar can be dissolved in the same amount of water. Once dissolved, however, the sugar will raise the boiling point of the water. The result of these physical chemistry properties is a specific relationship between the solution's boiling point and the amount of sugar solids contained therein. The first part of basic candy making, therefore, is cooking a sugar solution to a specific temperature to form a supersaturated solution with known solids content. When this solution cools, the sugar's solubility decreases and the sugar crystallizes out of solution. The next part of making a candy is controlling how this recrystallization takes place. It is also known as graining. Varying how the supersaturated syrup is physically treated while it cools controls recrystallization. -
Fake Glass Bottle / Sugar Glass
FAKE GLASS BOTTLE / SUGAR GLASS Step 1: What you are going to need. Materials for the mold: Oomoo #25 or 30 Mold release Glass bottle to be molded Plastic bottle ( at least 1" wider than the bottle to be mold) Materials for the candy glass: Sugar Light corn syrup Cream of tartar Water Food colorant Tools: Candy thermometer An old cooking pot Thick gloves Step 2: Prepare the bottle and mold Prepare the bottle: First you need to remove the stickers and labels from your bottle, save this so you can put them back on your fake bottle for a more realistic look. Clean the bottle really good, make sure there is no residue of any can because any of this will be transfer to the mold and the to you fake bottle. After the bottle is clean apply the mold release following the instruction on the can, in my case i only had to spray a thin coat all around. Make the mold: Get your big plastic bottle ( it should be at least 2" taller than the glass bottle and 1" wider ) Cut the top of the bottle, apply mold release to the interior of the plastic bottle. Hang the glass bottle inside the plastic bottle it should have 1/2" of free space all around and 1" in the bottom and 1" in the top. TIP: is a good idea to fill the glass bottle with water that way it will not float when you start pouring the silicone mix) Pour the mix really slowly with out disturbing the glass bottle cover at least 1/2" over the top of the bottle. -

Hard Candy: from Hard Crack to the Glass Transition
Hard Candy: from Hard Crack to the Glass Transition Careful control over formulation and processing conditions is required to produce quality hard candy with good shelf life. Richard W. Hartel, PhD University of Wisconsin–Madison ard candy is produced by boiling a mass turns into a sugar glass with very low Hsugar syrup to the hard crack tem- molecular mobility. It is this low mobility perature which, on a candy thermometer, that provides the long stability and shelf life is about 149°C (300°F). At hard crack, water of hard candy, as long as proper storage con- content is low and the sugar mass is ditions (cool and dry) are maintained. extremely viscous. After flavoring, the candy Besides flavor and color, hard candy for- mass is either deposited into molds or mulations vary primarily in their saccharide cooled to a highly plastic state that can be profile, with the ratio of sucrose to glucose Richard Hartel is a easily formed into desirable shapes. Upon syrup solids varying from as high as 80:20 down professor of food cooling to room temperature, the piece sets to about 30:70 (although a more common engineering in the range might be 60:40 to 40:60), as seen in Fig- into a sugar glass that, if protected from Food Science depart- ure 1. A typical reducing sugar target is 16 to ment at the University heat and humidity, retains its quality for of Wisconsin –Madi- years. After touching on the basics behind son. He also is course hard candy manufacture, this review will Typical Compositions (in %) of Fiinished Hard Candy coordinator for the discuss the numerous chemical and physical two-week Resident changes that occur in the sugar mass dur- High Medium Low Course in Candy Sucrose Sucrose Sucrose Technology held ing the manufacturing process that can Sucrose 70 – 80 50 – 60 30 – 40 annually in Madison.