GIGSEA: Genotype Imputed Gene Set Enrichment Analysis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Allele-Specific Expression of Ribosomal Protein Genes in Interspecific Hybrid Catfish
Allele-specific Expression of Ribosomal Protein Genes in Interspecific Hybrid Catfish by Ailu Chen A dissertation submitted to the Graduate Faculty of Auburn University in partial fulfillment of the requirements for the Degree of Doctor of Philosophy Auburn, Alabama August 1, 2015 Keywords: catfish, interspecific hybrids, allele-specific expression, ribosomal protein Copyright 2015 by Ailu Chen Approved by Zhanjiang Liu, Chair, Professor, School of Fisheries, Aquaculture and Aquatic Sciences Nannan Liu, Professor, Entomology and Plant Pathology Eric Peatman, Associate Professor, School of Fisheries, Aquaculture and Aquatic Sciences Aaron M. Rashotte, Associate Professor, Biological Sciences Abstract Interspecific hybridization results in a vast reservoir of allelic variations, which may potentially contribute to phenotypical enhancement in the hybrids. Whether the allelic variations are related to the downstream phenotypic differences of interspecific hybrid is still an open question. The recently developed genome-wide allele-specific approaches that harness high- throughput sequencing technology allow direct quantification of allelic variations and gene expression patterns. In this work, I investigated allele-specific expression (ASE) pattern using RNA-Seq datasets generated from interspecific catfish hybrids. The objective of the study is to determine the ASE genes and pathways in which they are involved. Specifically, my study investigated ASE-SNPs, ASE-genes, parent-of-origins of ASE allele and how ASE would possibly contribute to heterosis. My data showed that ASE was operating in the interspecific catfish system. Of the 66,251 and 177,841 SNPs identified from the datasets of the liver and gill, 5,420 (8.2%) and 13,390 (7.5%) SNPs were identified as significant ASE-SNPs, respectively. -

Supplementary Figures 1-14 and Supplementary References
SUPPORTING INFORMATION Spatial Cross-Talk Between Oxidative Stress and DNA Replication in Human Fibroblasts Marko Radulovic,1,2 Noor O Baqader,1 Kai Stoeber,3† and Jasminka Godovac-Zimmermann1* 1Division of Medicine, University College London, Center for Nephrology, Royal Free Campus, Rowland Hill Street, London, NW3 2PF, UK. 2Insitute of Oncology and Radiology, Pasterova 14, 11000 Belgrade, Serbia 3Research Department of Pathology and UCL Cancer Institute, Rockefeller Building, University College London, University Street, London WC1E 6JJ, UK †Present Address: Shionogi Europe, 33 Kingsway, Holborn, London WC2B 6UF, UK TABLE OF CONTENTS 1. Supplementary Figures 1-14 and Supplementary References. Figure S-1. Network and joint spatial razor plot for 18 enzymes of glycolysis and the pentose phosphate shunt. Figure S-2. Correlation of SILAC ratios between OXS and OAC for proteins assigned to the SAME class. Figure S-3. Overlap matrix (r = 1) for groups of CORUM complexes containing 19 proteins of the 49-set. Figure S-4. Joint spatial razor plots for the Nop56p complex and FIB-associated complex involved in ribosome biogenesis. Figure S-5. Analysis of the response of emerin nuclear envelope complexes to OXS and OAC. Figure S-6. Joint spatial razor plots for the CCT protein folding complex, ATP synthase and V-Type ATPase. Figure S-7. Joint spatial razor plots showing changes in subcellular abundance and compartmental distribution for proteins annotated by GO to nucleocytoplasmic transport (GO:0006913). Figure S-8. Joint spatial razor plots showing changes in subcellular abundance and compartmental distribution for proteins annotated to endocytosis (GO:0006897). Figure S-9. Joint spatial razor plots for 401-set proteins annotated by GO to small GTPase mediated signal transduction (GO:0007264) and/or GTPase activity (GO:0003924). -

1 AGING Supplementary Table 2
SUPPLEMENTARY TABLES Supplementary Table 1. Details of the eight domain chains of KIAA0101. Serial IDENTITY MAX IN COMP- INTERFACE ID POSITION RESOLUTION EXPERIMENT TYPE number START STOP SCORE IDENTITY LEX WITH CAVITY A 4D2G_D 52 - 69 52 69 100 100 2.65 Å PCNA X-RAY DIFFRACTION √ B 4D2G_E 52 - 69 52 69 100 100 2.65 Å PCNA X-RAY DIFFRACTION √ C 6EHT_D 52 - 71 52 71 100 100 3.2Å PCNA X-RAY DIFFRACTION √ D 6EHT_E 52 - 71 52 71 100 100 3.2Å PCNA X-RAY DIFFRACTION √ E 6GWS_D 41-72 41 72 100 100 3.2Å PCNA X-RAY DIFFRACTION √ F 6GWS_E 41-72 41 72 100 100 2.9Å PCNA X-RAY DIFFRACTION √ G 6GWS_F 41-72 41 72 100 100 2.9Å PCNA X-RAY DIFFRACTION √ H 6IIW_B 2-11 2 11 100 100 1.699Å UHRF1 X-RAY DIFFRACTION √ www.aging-us.com 1 AGING Supplementary Table 2. Significantly enriched gene ontology (GO) annotations (cellular components) of KIAA0101 in lung adenocarcinoma (LinkedOmics). Leading Description FDR Leading Edge Gene EdgeNum RAD51, SPC25, CCNB1, BIRC5, NCAPG, ZWINT, MAD2L1, SKA3, NUF2, BUB1B, CENPA, SKA1, AURKB, NEK2, CENPW, HJURP, NDC80, CDCA5, NCAPH, BUB1, ZWILCH, CENPK, KIF2C, AURKA, CENPN, TOP2A, CENPM, PLK1, ERCC6L, CDT1, CHEK1, SPAG5, CENPH, condensed 66 0 SPC24, NUP37, BLM, CENPE, BUB3, CDK2, FANCD2, CENPO, CENPF, BRCA1, DSN1, chromosome MKI67, NCAPG2, H2AFX, HMGB2, SUV39H1, CBX3, TUBG1, KNTC1, PPP1CC, SMC2, BANF1, NCAPD2, SKA2, NUP107, BRCA2, NUP85, ITGB3BP, SYCE2, TOPBP1, DMC1, SMC4, INCENP. RAD51, OIP5, CDK1, SPC25, CCNB1, BIRC5, NCAPG, ZWINT, MAD2L1, SKA3, NUF2, BUB1B, CENPA, SKA1, AURKB, NEK2, ESCO2, CENPW, HJURP, TTK, NDC80, CDCA5, BUB1, ZWILCH, CENPK, KIF2C, AURKA, DSCC1, CENPN, CDCA8, CENPM, PLK1, MCM6, ERCC6L, CDT1, HELLS, CHEK1, SPAG5, CENPH, PCNA, SPC24, CENPI, NUP37, FEN1, chromosomal 94 0 CENPL, BLM, KIF18A, CENPE, MCM4, BUB3, SUV39H2, MCM2, CDK2, PIF1, DNA2, region CENPO, CENPF, CHEK2, DSN1, H2AFX, MCM7, SUV39H1, MTBP, CBX3, RECQL4, KNTC1, PPP1CC, CENPP, CENPQ, PTGES3, NCAPD2, DYNLL1, SKA2, HAT1, NUP107, MCM5, MCM3, MSH2, BRCA2, NUP85, SSB, ITGB3BP, DMC1, INCENP, THOC3, XPO1, APEX1, XRCC5, KIF22, DCLRE1A, SEH1L, XRCC3, NSMCE2, RAD21. -

MRPL24 Polyclonal Antibody
PRODUCT DATA SHEET Bioworld Technology,Inc. MRPL24 polyclonal antibody Catalog: BS65388 Host: Rabbit Reactivity: Human,Mouse,Ra BackGround: term. Avoid freeze-thaw cycles. mitochondrial ribosomal protein L24(MRPL24) Homo Specificity: sapiens Mammalian mitochondrial ribosomal proteins are MRP-L24 Polyclonal Antibody detects endogenous levels encoded by nuclear genes and help in protein synthesis of MRP-L24 protein. within the mitochondrion. Mitochondrial ribosomes (mi- DATA: toribosomes) consist of a small 28S subunit and a large 39S subunit. They have an estimated 75% protein to rRNA composition compared to prokaryotic ribosomes, where this ratio is reversed. Another difference between mammalian mitoribosomes and prokaryotic ribosomes is that the latter contain a 5S rRNA. Among different spe- cies, the proteins comprising the mitoribosome differ greatly in sequence, and sometimes in biochemical prop- Western Blot analysis of various cells using MRP-L24 Polyclonal Anti- erties, which prevents easy recognition by sequence ho- body diluted at 1:2000 mology. This gene encodes a 39S subunit protein which is more than twice the size of its E.coli counterpart (EcoL24). Sequence analysis identified two transcript variants that encode the same protein. [provided by Ref- Seq, Jul 2008], Product: Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide. Immunohistochemistry analysis of paraffin-embedded human breast Molecular Weight: carcinoma tissue, using MRPL24 Antibody. The picture on the right is ~ 32 kDa blocked with the synthesized peptide. Swiss-Prot: Q96A35 Purification&Purity: The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific im- munogen. Applications: Western blot analysis of lysates from HUVEC cells, using MRPL24 An- Western Blot: 1/500 - 1/2000. -

YEASTRACT - Genes Grouped by TF, Ordered by the Percentage of Genes Regulated by TF, Relative to the Total Number of Genes in the List 08/07/14 16:17
YEASTRACT - Genes grouped by TF, ordered by the percentage of genes regulated by TF, relative to the total number of genes in the list 08/07/14 16:17 Home > Group Genes by TF > Result Contact Us - Tutorial - Tutorial Genes grouped by TF, ordered by the percentage of genes regulated by TF, relative to the total number of genes in the list Quick search... Search Documented regulations suported by direct or indirect or undefined evidence. Unknown gene/ORF name(s), 'YFL013W-A'. DISCOVERER Transcription Transcriptional Regulatory % ORF/Genes Regulatory Associations: Factor Network - Search for TFs NTG1 YAL045c HAP3 - Search for Genes ATP1 YBL107c ALG1 - Search for Associations YSA1 CNS1 PDB1 TSC10 RER1 YCL049c PRD1 Group genes: AHC2 GET3 BPL1 - Group by TF YDL144c GCV1 PST2 - Group by GO PST1 SED1 YDR262w YDR266c MSW1 FRQ1 Pattern Matching: TSA2 STP1 PHO8 RAD23 - Search by DNA Motif YEL047c CAN1 YEL074w - Find TF Binding Site(s) PMI40 FMP52 CEM1 - Search Motifs on Motifs SER3 SHC1 YER130c Utilities: PDA1 YER189w AGP3 - ORF List ⇔ Gene List DUG1 CDH1 OCH1 - IUPAC Code Generation YGL114w MRM2 CHO2 - Generate Regulation Matrix PMT6 TRX2 YSC84 CHS7 Yap1p 24.9 % YSP1 FAA3 LYS1 GTT1 Retrieve: YJL045w GZF3 LCB3 - Transcription Factors List HXT9 VPS55 HOM6 BAT2 - Upstream Sequence THI11 RSM22 UBA1 - Flat files RHO4 PAM17 YKR070w YKR077w GTT2 YLL067c About Yeastract: ALT1 HOG1 MAS1 - Contact Us ECM38 ILV5 MRPL4 - Cite YEASTRACT TSA1 PRE8 TAF13 VAN1 - Acknowledgments YTA12 NUP53 SPG5 - Credits MRPL24 NIS1 APP1 CPT1 FMP41 MRPL19 YNL208w CWC25 LYP1 TOF1 -

Supplementary Dataset S2
mitochondrial translational termination MRPL28 MRPS26 6 MRPS21 PTCD3 MTRF1L 4 MRPL50 MRPS18A MRPS17 2 MRPL20 MRPL52 0 MRPL17 MRPS33 MRPS15 −2 MRPL45 MRPL30 MRPS27 AURKAIP1 MRPL18 MRPL3 MRPS6 MRPS18B MRPL41 MRPS2 MRPL34 GADD45GIP1 ERAL1 MRPL37 MRPS10 MRPL42 MRPL19 MRPS35 MRPL9 MRPL24 MRPS5 MRPL44 MRPS23 MRPS25 ITB ITB ITB ITB ICa ICr ITL original ICr ICa ITL ICa ITL original ICr ITL ICr ICa mitochondrial translational elongation MRPL28 MRPS26 6 MRPS21 PTCD3 MRPS18A 4 MRPS17 MRPL20 2 MRPS15 MRPL45 MRPL52 0 MRPS33 MRPL30 −2 MRPS27 AURKAIP1 MRPS10 MRPL42 MRPL19 MRPL18 MRPL3 MRPS6 MRPL24 MRPS35 MRPL9 MRPS18B MRPL41 MRPS2 MRPL34 MRPS5 MRPL44 MRPS23 MRPS25 MRPL50 MRPL17 GADD45GIP1 ERAL1 MRPL37 ITB ITB ITB ITB ICa ICr original ICr ITL ICa ITL ICa ITL original ICr ITL ICr ICa translational termination MRPL28 MRPS26 6 MRPS21 PTCD3 C12orf65 4 MTRF1L MRPL50 MRPS18A 2 MRPS17 MRPL20 0 MRPL52 MRPL17 MRPS33 −2 MRPS15 MRPL45 MRPL30 MRPS27 AURKAIP1 MRPL18 MRPL3 MRPS6 MRPS18B MRPL41 MRPS2 MRPL34 GADD45GIP1 ERAL1 MRPL37 MRPS10 MRPL42 MRPL19 MRPS35 MRPL9 MRPL24 MRPS5 MRPL44 MRPS23 MRPS25 ITB ITB ITB ITB ICa ICr original ICr ITL ICa ITL ICa ITL original ICr ITL ICr ICa translational elongation DIO2 MRPS18B MRPL41 6 MRPS2 MRPL34 GADD45GIP1 4 ERAL1 MRPL37 2 MRPS10 MRPL42 MRPL19 0 MRPL30 MRPS27 AURKAIP1 −2 MRPL18 MRPL3 MRPS6 MRPS35 MRPL9 EEF2K MRPL50 MRPS5 MRPL44 MRPS23 MRPS25 MRPL24 MRPS33 MRPL52 EIF5A2 MRPL17 SECISBP2 MRPS15 MRPL45 MRPS18A MRPS17 MRPL20 MRPL28 MRPS26 MRPS21 PTCD3 ITB ITB ITB ITB ICa ICr ICr ITL original ITL ICa ICa ITL ICr ICr ICa original -

A Homozygous MRPL24 Mutation Causes a Complex Movement Disorder and Affects the Mitoribosome Assembly T
Neurobiology of Disease 141 (2020) 104880 Contents lists available at ScienceDirect Neurobiology of Disease journal homepage: www.elsevier.com/locate/ynbdi A homozygous MRPL24 mutation causes a complex movement disorder and affects the mitoribosome assembly T Michela Di Nottiaa,1, Maria Marcheseb,1, Daniela Verrignia, Christian Daniel Muttic, Alessandra Torracoa, Romina Olivad, Erika Fernandez-Vizarrac, Federica Moranib, Giulia Trania, Teresa Rizzaa, Daniele Ghezzie,f, Anna Ardissoneg,h, Claudia Nestib, Gessica Vascoi, Massimo Zevianic, Michal Minczukc, Enrico Bertinia, Filippo Maria Santorellib,2, ⁎ Rosalba Carrozzoa, ,2 a Unit of Muscular and Neurodegenerative Disorders, Laboratory of Molecular Medicine, Bambino Gesù Children's Hospital, IRCCS, Rome, Italy b Molecular Medicine & Neurogenetics, IRCCS Fondazione Stella Maris, Pisa, Italy c MRC Mitochondrial Biology Unit, University of Cambridge, Cambridge, UK d Department of Sciences and Technologies, University Parthenope of Naples, Naples, Italy e Unit of Medical Genetics and Neurogenetics, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy f Department of Pathophysiology and Transplantation, University of Milan, Milan, Italy g Child Neurology Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy h Department of Molecular and Translational Medicine DIMET, University of Milan-Bicocca, Milan, Italy i Department of Neurosciences, IRCCS Bambino Gesù Children Hospital, Rome, Italy ARTICLE INFO ABSTRACT Keywords: Mitochondrial ribosomal protein large 24 (MRPL24) is 1 of the 82 protein components of mitochondrial ribo- Mitochondrial disorders somes, playing an essential role in the mitochondrial translation process. Movement disorder We report here on a baby girl with cerebellar atrophy, choreoathetosis of limbs and face, intellectual dis- MRPL24 ability and a combined defect of complexes I and IV in muscle biopsy, caused by a homozygous missense mu- Mitoribosomes tation identified in MRPL24. -

Transcriptomic and Proteomic Landscape of Mitochondrial
TOOLS AND RESOURCES Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals Inge Ku¨ hl1,2†*, Maria Miranda1†, Ilian Atanassov3, Irina Kuznetsova4,5, Yvonne Hinze3, Arnaud Mourier6, Aleksandra Filipovska4,5, Nils-Go¨ ran Larsson1,7* 1Department of Mitochondrial Biology, Max Planck Institute for Biology of Ageing, Cologne, Germany; 2Department of Cell Biology, Institute of Integrative Biology of the Cell (I2BC) UMR9198, CEA, CNRS, Univ. Paris-Sud, Universite´ Paris-Saclay, Gif- sur-Yvette, France; 3Proteomics Core Facility, Max Planck Institute for Biology of Ageing, Cologne, Germany; 4Harry Perkins Institute of Medical Research, The University of Western Australia, Nedlands, Australia; 5School of Molecular Sciences, The University of Western Australia, Crawley, Australia; 6The Centre National de la Recherche Scientifique, Institut de Biochimie et Ge´ne´tique Cellulaires, Universite´ de Bordeaux, Bordeaux, France; 7Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm, Sweden Abstract Dysfunction of the oxidative phosphorylation (OXPHOS) system is a major cause of human disease and the cellular consequences are highly complex. Here, we present comparative *For correspondence: analyses of mitochondrial proteomes, cellular transcriptomes and targeted metabolomics of five [email protected] knockout mouse strains deficient in essential factors required for mitochondrial DNA gene (IKu¨ ); expression, leading to OXPHOS dysfunction. Moreover, -

GIGSEA: Genotype Imputed Gene Set Enrichment Analysis
GIGSEA: Genotype Imputed Gene Set Enrichment Analysis Shijia Zhu Department of Genetics and Genomic Sciences and Icahn Institute for Genomics and Multiscale Biology, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA August 28, 2017 Contents Abstract . .1 1. Import packages . .1 2. Quick start . .2 3. One example of MetaXcan output . .2 4. Load data of gene sets . .4 4.1 Discrete-valued gene sets: . .4 4.2 Continuous-valued gene sets: . .6 4.3 User self-defined gene set . .7 5. Gene set enrichment analysis . .9 5.1 Gene set enrichment analysis using weighted simple linear regression . .9 5.2 Gene set enrichment analysis using weighted multiple regression model . 10 5.3 One-step weightedGSEA . 11 Abstract We presented the Genotype Imputed Gene Set Enrichment Analysis (GIGSEA), a novel method that uses GWAS-and-eQTL-imputed differential gene expression to interrogate gene set enrichment for the trait-associated SNPs. By incorporating eQTL from large gene expression studies, e.g. GTEx, GIGSEA appropriately addresses such challenges for SNP enrichment as gene size, gene boundary, andmultiple-marker regulation. The weighted linear regression model, taking as weights both imputation accuracy and model completeness, was used to test the enrichment, properly adjusting the bias due to redundancy in different gene sets. The permutation test, furthermore, is used to evaluate the significance of enrichment, whose efficiency can be largely elevated by expressing the computational intensive part in terms of large matrix operation. We have shown the appropriate type I error rates for GIGSEA (<5%), and the preliminary results also demonstrate its good performance to uncover the real biological signal. -

Cardiomyopathy Is Associated with Ribosomal Protein Gene Haploinsufficiency In
Genetics: Published Articles Ahead of Print, published on September 2, 2011 as 10.1534/genetics.111.131482 Cardiomyopathy is associated with ribosomal protein gene haploinsufficiency in Drosophila melanogaster Michelle E. Casad*, Dennis Abraham§, Il-Man Kim§, Stephan Frangakis*, Brian Dong*, Na Lin§1, Matthew J. Wolf§, and Howard A. Rockman*§** Department of *Cell Biology, §Medicine, and **Molecular Genetics and Microbiology, Duke University Medical Center, Durham, NC 27710 1Current Address: Institute of Molecular Medicine, Peking University, Beijing, China, 100871 1 Copyright 2011. Running Title: Cardiomyopathy in Minute Drosophila Key Words: Drosophila Minute syndrome Cardiomyopathy Address for correspondence: Howard A. Rockman, M.D. Department of Medicine Duke University Medical Center, DUMC 3104 226 CARL Building, Research Drive Durham, NC, 27710 Tel: (919) 668-2520 FAX: (919) 668-2524 Email: [email protected] 2 ABSTRACT The Minute syndrome in Drosophila melanogaster is characterized by delayed development, poor fertility, and short slender bristles. Many Minute loci correspond to disruptions of genes for cytoplasmic ribosomal proteins, and therefore the phenotype has been attributed to alterations in translational processes. Although protein translation is crucial for all cells in an organism, it is unclear why Minute mutations cause effects in specific tissues. To determine whether the heart is sensitive to haploinsufficiency of genes encoding ribosomal proteins, we measured heart function of Minute mutants using optical coherence tomography. We found that cardiomyopathy is associated with the Minute syndrome caused by haploinsufficiency of genes encoding cytoplasmic ribosomal proteins. While mutations of genes encoding non-Minute cytoplasmic ribosomal proteins are homozygous lethal, heterozygous deficiencies spanning these non-Minute genes did not cause a change in cardiac function. -

MRPL24 Rabbit Pab
Leader in Biomolecular Solutions for Life Science MRPL24 Rabbit pAb Catalog No.: A4967 1 Publications Basic Information Background Catalog No. Mammalian mitochondrial ribosomal proteins are encoded by nuclear genes and help in A4967 protein synthesis within the mitochondrion. Mitochondrial ribosomes (mitoribosomes) consist of a small 28S subunit and a large 39S subunit. They have an estimated 75% Observed MW protein to rRNA composition compared to prokaryotic ribosomes, where this ratio is 25kDa reversed. Another difference between mammalian mitoribosomes and prokaryotic ribosomes is that the latter contain a 5S rRNA. Among different species, the proteins Calculated MW comprising the mitoribosome differ greatly in sequence, and sometimes in biochemical 24kDa properties, which prevents easy recognition by sequence homology. This gene encodes a 39S subunit protein which is more than twice the size of its E.coli counterpart (EcoL24). Category Sequence analysis identified two transcript variants that encode the same protein. Primary antibody Applications WB Cross-Reactivity Human, Mouse, Rat Recommended Dilutions Immunogen Information WB 1:500 - 1:2000 Gene ID Swiss Prot 79590 Q96A35 Immunogen Recombinant fusion protein containing a sequence corresponding to amino acids 1-216 of human MRPL24 (NP_078816.2). Synonyms MRPL24;L24mt;MRP-L18;MRP-L24 Contact Product Information www.abclonal.com Source Isotype Purification Rabbit IgG Affinity purification Storage Store at -20℃. Avoid freeze / thaw cycles. Buffer: PBS with 0.02% sodium azide,50% glycerol,pH7.3. Validation Data Western blot analysis of extracts of various cell lines, using MRPL24 antibody (A4967) at 1:1000 dilution. Secondary antibody: HRP Goat Anti-Rabbit IgG (H+L) (AS014) at 1:10000 dilution. -
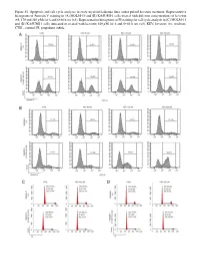
Figure S1. Apoptosis and Cell Cycle Analyses in Acute Myeloid Leukemia Lines Under Pulsed Kevetrin Treatment
Figure S1. Apoptosis and cell cycle analyses in acute myeloid leukemia lines under pulsed kevetrin treatment. Representative histograms of Annexin V staining in (A) MOLM‑13 and (B) KASUMI‑1 cells treated with different concentrations of kevetrin (85, 170 and 340 µM) for 6 and 6+66 h wo (x3). Representative histograms of PI staining for cell cycle analysis in (C) MOLM‑13 and (D) KASUMI‑1 cells untreated or treated with kevetrin 340 µM for 6 and 6+66 h wo (x3). KEV, kevetrin; wo, washout; CTRL, control; PI, propidium iodide. Figure S2. Viability of acute myeloid leukemia cell lines after 72 h of kevetrin treatment. Viability of OCI‑AML3, MOLM‑13, KASUMI‑1 and NOMO‑1 cell lines treated with different concentrations of kevetrin (85, 170 and 340 µM) for 72 h. Values represent the mean ± standard deviation of three biological replicates. **P<0.01, ***P<0.001. Figure S3. Apoptosis analysis in acute myeloid leukemia cell lines under 24 h of continuous kevetrin treatment. Representative histograms of Annexin V staining in OCI‑AML3, MOLM‑13, KASUMI‑1 and NOMO‑1 cells treated with different concentra‑ tions of kevetrin (85, 170 and 340 µM) for 24 h. KEV, kevetrin; CTRL, control. Figure S4. Apoptosis analysis in acute myeloid leukemia cell lines under 48 h of continuous kevetrin treatment. Representative histograms of Annexin V staining in OCI‑AML3, MOLM‑13, KASUMI‑1 and NOMO‑1 cells treated with different concentra‑ tions of kevetrin (85, 170 and 340 µM) for 48 h. KEV, kevetrin; CTRL, control. Figure S5. Apoptosis analyses in acute myeloid leukemia cell lines under continuous kevetrin treatment.