Evolutionary Dynamics of Host Specialization in Wood-Decay Fungi
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
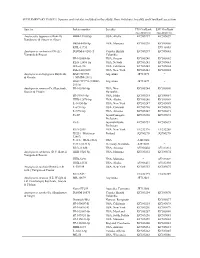
Suppl Table I
SUPPLEMENTARY TABLE I. Species and isolates included in the study, their vouchers, locality and GenBank accession Species Isolate number Locality ITS GenBank LSU GenBank accession no. accession no. Amylocystis lapponica (Romell) HHB-13400-Sp USA, Alaska KC585237 KC585059 Bondartsev & Singer ex Singer OKM-4418-Sp USA, Montana KC585238 KC585060 KHL-11755 − EU118603 Amyloporia carbonica (Overh.) DAOM-F-8281-T Canada, British KC585239 KC585061 Vampola & Pouzar Columbia FP-105585-Sp USA, Oregon KC585240 KC585062 RLG-12496-Sp USA, Nevada KC585241 KC585063 Wilcox-96 USA, California KC585242 KC585064 Zabel-40-GLN USA, New York KC585243 KC585065 Amyloporia nothofaginea Rajchenb. BAFC519794 Argentina JF713078 − & Gorjón (=MMBP-2011) BAFC519796 (=MBP- Argentina JF713079 − 2011a) Amyloporia sinuosa (Fr.) Rajchenb., FP-105386-Sp USA, New KC585244 KC585066 Gorjón & Pildain Hampshire FP-94464-Sp USA, Idaho KC585245 KC585067 HHB-12878-Sp USA, Alaska KC585246 KC585068 L-14130-Sp USA, New York KC585247 KC585069 L-6192-Sp USA, Colorado KC585248 KC585070 L-9792-Sp USA, Arizona KC585249 KC585071 Pa-3C JapanYamagata KC585250 KC585072 Prefecture Pa-3e JapanShizuoka KC585251 KC585073 Prefecture RLG-2538 USA, New York EU232196 EU232288 X725 (=Miettinen- Finland JQ700270 JQ700270 12407) P-115 (=RLG-2538) USA AJ416068 − P-211 (G-214) Germany, Karlsrube AJ345011 − RLG-1182R USA, Arizona AY966450 AY333831 Amyloporia sitchensis (D.V. Baxter) HHB-5320-Sp USA, Montana KC585252 KC585074 Vampola & Pouzar HHB-5298 USA, Montana − AY333829 HHB-12513 USA, Alaska AY966451 AY333830 -

Comparison of Wood Decay Among Diverse Lignicolous Fungi
Mycologia, 89(2), 1997, pp. 199-219. ? 1997 by The New York Botanical Garden, Bronx, NY 10458-5126 Comparison of wood decay among diverse lignicolous fungi James J. Worrall 1 orders of gasteromycetes. Selective delignification Susan E. Anagnost was most pronounced at low weight losses. Certain Robert A. Zabel decay features similar to those in the Ascomycota Collegeof EnvironmentalScience and Forestry,State were found in the Auriculariales, consistent with hy- Universityof New York,Syracuse, New York13210 potheses that place that order near the phylogenetic root of Basidiomycota. A sequence of origins of decay types is proposed. Abstract: In decay tests with 98 isolates (78 species) Key Words: Agaricales, Aphyllophorales, Auricu- of lignicolous fungi followed by chemical and ana- lariales, brown rot, Dacrymycetales, Exidiaceae, phy- tomical analyses, the validity of the generally accept- logeny, soft rot, white rot, Xylariales ed, major decay types (white, brown, and soft rot) was confirmed, and no new major types proposed. We could soft rot from other distinguish decay types INTRODUCTION based on anatomical and chemical criteria, without reliance on cavities or recourse to taxonomy of causal Concepts of wood decay continue to be based agents. Chemically, soft rot of birch could be distin- largely on a limited range of lignicolous fungi, pri- guished from white rot by lower Klason lignin loss, marily in the families Polyporaceae, Hymenochaeta- and from brown rot by much lower alkali solubility. ceae and Corticiaceae sensu lato of the order -

A New Benzoquinone and a New Benzofuran from the Edible
Food Chemistry 141 (2013) 1614–1618 Contents lists available at SciVerse ScienceDirect Food Chemistry journal homepage: www.elsevier.com/locate/foodchem A new benzoquinone and a new benzofuran from the edible mushroom Neolentinus lepideus and their inhibitory activity in NO production inhibition assay ⇑ ⇑ Yongxia Li a,b,1, Li Bao a,1, Bin Song c, Junjie Han b, Heran Li b, , Feng Zhao d, Hongwei Liu a, a State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, No. 9, Beiertiao, Zhongguancun, Haidian District, Beijing 100190, People’s Republic of China b College of Pharmacy, Soochow University, No. 199, Ren Ai Rd., Suzhou Industrial Park, Suzhou, People’s Republic of China c Guangdong Institute of Microbiology, Guangdong Academy of Sciences, No. 100, Xianlie Road, Yuexiu District, Guangdong 510070, People’s Republic of China d School of Pharmacy, Yantai University, No. 32, Qingquan Road, Laishan District, Yantai 264005, People’s Republic of China article info abstract Article history: The fruiting bodies or mycelia of mushrooms have been used as food and food-flavoring material for cen- Received 16 October 2012 turies due to their nutritional and medicinal value and the diversity of their bioactive components. The Received in revised form 21 February 2013 present research is the first to investigate the bioactive secondary metabolites from the solid culture of Accepted 30 April 2013 the edible mushroom Neolentinus lepideus. Two new secondary metabolites, 5-methoxyisobenzofuran- Available online 23 May 2013 4,7(1H,3H)-dione (1) and 1,3-dihydroisobenzofuran-4,6-diol (2), as well as seven known compounds including one benzoquinone derivative (3) and six cinnamic acid derivatives (4–9) were obtained. -

Basidiomycota: Agaricales) Introducing the Ant-Associated Genus Myrmecopterula Gen
Leal-Dutra et al. IMA Fungus (2020) 11:2 https://doi.org/10.1186/s43008-019-0022-6 IMA Fungus RESEARCH Open Access Reclassification of Pterulaceae Corner (Basidiomycota: Agaricales) introducing the ant-associated genus Myrmecopterula gen. nov., Phaeopterula Henn. and the corticioid Radulomycetaceae fam. nov. Caio A. Leal-Dutra1,5, Gareth W. Griffith1* , Maria Alice Neves2, David J. McLaughlin3, Esther G. McLaughlin3, Lina A. Clasen1 and Bryn T. M. Dentinger4 Abstract Pterulaceae was formally proposed to group six coralloid and dimitic genera: Actiniceps (=Dimorphocystis), Allantula, Deflexula, Parapterulicium, Pterula, and Pterulicium. Recent molecular studies have shown that some of the characters currently used in Pterulaceae do not distinguish the genera. Actiniceps and Parapterulicium have been removed, and a few other resupinate genera were added to the family. However, none of these studies intended to investigate the relationship between Pterulaceae genera. In this study, we generated 278 sequences from both newly collected and fungarium samples. Phylogenetic analyses supported with morphological data allowed a reclassification of Pterulaceae where we propose the introduction of Myrmecopterula gen. nov. and Radulomycetaceae fam. nov., the reintroduction of Phaeopterula, the synonymisation of Deflexula in Pterulicium, and 53 new combinations. Pterula is rendered polyphyletic requiring a reclassification; thus, it is split into Pterula, Myrmecopterula gen. nov., Pterulicium and Phaeopterula. Deflexula is recovered as paraphyletic alongside several Pterula species and Pterulicium, and is sunk into the latter genus. Phaeopterula is reintroduced to accommodate species with darker basidiomes. The neotropical Myrmecopterula gen. nov. forms a distinct clade adjacent to Pterula, and most members of this clade are associated with active or inactive attine ant nests. -

Gymnosperms) of New York State
QK 129 . C667 1992 Pinophyta (Gymnosperms) of New York State Edward A. Cope The L. H. Bailey Hortorium Cornell University Contributions to a Flora of New York State IX Richard S. Mitchell, Editor 1992 Bulletin No. 483 New York State Museum The University of the State of New York THE STATE EDUCATION DEPARTMENT Albany, New York 12230 V A ThL U: ESTHER T. SVIERTZ LIBRARY THI-: ?‘HW YORK BOTANICAL GARDEN THE LuESTHER T. MERTZ LIBRARY THE NEW YORK BOTANICAL GARDEN Pinophyta (Gymnosperms) of New York State Edward A. Cope The L. H. Bailey Hortorium Cornell University Contributions to a Flora of New York State IX Richard S. Mitchell, Editor 1992 Bulletin No. 483 New York State Museum The University of the State of New York THE STATE EDUC ATION DEPARTMENT Albany, New York 12230 THE UNIVERSITY OF THE STATE OF NEW YORK Regents of The University Martin C. Barell, Chancellor, B.A., I.A., LL.B. Muttontown R. Carlos Carballada, Vice Chancellor, B.S. Rochester Willard A. Genrich, LL.B. Buffalo Emlyn I. Griffith. A.B.. J.D. Rome Jorge L. Batista, B.A.. J.D. Bronx Laura Bradley Chodos, B.A., M.A. Vischer Ferry Louise P. Matteoni, B.A., M.A., Ph.D. Bayside J. Edward Meyer, B.A., LL.B. Chappaqua FloydS. Linton, A.B., M.A., M.P.A. Miller Place Mimi Levin Lif.ber, B.A., M.A. Manhattan Shirley C. Brown, B.A., M.A., Ph.D. Albany Norma Gluck, B.A., M.S.W. Manhattan Adelaide L. Sanford, B.A., M.A., P.D. -

(2014), Volume 2, Issue 11, 238-245
ISSN 2320-5407 International Journal of Advanced Research (2014), Volume 2, Issue 11, 238-245 Journal homepage: http://www.journalijar.com INTERNATIONAL JOURNAL OF ADVANCED RESEARCH RESEARCH ARTICLE Diversity of family Meruliaceae from Jammu Division (J&K), India Jyoti*, Avneet Pal Singh & Gurpaul Singh Dhingra Department of Botany, Punjabi University, Patiala, 147002 India Manuscript Info Abstract Manuscript History: An account of eight resupinate, non-poroid taxa (Crustoderma corneum, Gyrophanopsis polonensis, Hyphoderma argillaceum, H. hjortstamii, H. Received: 25 September 2014 Final Accepted: 19 October 2014 setigerum, H. setigerum var. bicystidium, Hypochnicium wakefieldiae, Published Online: November 2014 Radulodon indicus) of family Meruliaceae (Class- Agaricomycetes, Phylum- Basidiomycota) has been given. All these are new reports for the Key words: Jammu Division in the state of Jammu and Kashmir (J&K). Of these, Basidiomycota, Agaricomycetes, Hyphoderma hjortstamii is a new record for India, Hypochnicium Meruliaceae. wakefieldiae new for the North Western Himalaya, Crustoderma corneum, Gyrophanopsis polonensis and H. setigerum var. bicystidium new for J&K. *Corresponding Author Jyoti Sharma Copy Right, IJAR, 2014,. All rights reserved Introduction While conducting fungal forays in the different localities of Jammu division in the state of Jammu and Kashmir (India), twelve collections of resupinate, non-poroid Agaricomycetous fungi were made. On the basis of comparison of macroscopic and microscopic features in the published literature (Thind & Rattan 1970, Eriksson & Ryvarden 1975, Rattan 1977, Eriksson & Ryvarden 1976, Eriksson et al. 1981, Wu SH. 1990, Stalpers 1998, Nakasone 2001, Bernicchia & Gorjón 2010), these have been identified as Crustoderma corneum, Gyrophanopsis polonensis, Hyphoderma argillaceum, H. hjortstamii, H. setigerum, H. setigerum var. bicystidium, Hypochnicium wakefieldiae and Radulodon indicus. -

Short-Read Sequencing for Genomic Analysis of the Brown Rot Fungus Fibroporia Radiculosa
Short-Read Sequencing for Genomic Analysis of the Brown Rot Fungus Fibroporia radiculosa Juliet D. Tang,a Andy D. Perkins,b Tad S. Sonstegard,c Steven G. Schroeder,c Shane C. Burgess,d and Susan V. Diehla Forest Products, Mississippi State University, Mississippi State, Mississippi, USAa; Computer Science and Engineering, Mississippi State University, Mississippi State, Mississippi, USAb; USDA ARS Bovine Functional Genomics Laboratory, Beltsville, Maryland, USAc; and Institute for Genomics, Biocomputing, and Biotechnology, Mississippi State University, Mississippi State, Mississippi, USAd The feasibility of short-read sequencing for genomic analysis was demonstrated for Fibroporia radiculosa, a copper-tolerant fungus that causes brown rot decay of wood. The effect of read quality on genomic assembly was assessed by filtering Illumina GAIIx reads from a single run of a paired-end library (75-nucleotide read length and 300-bp fragment size) at three different stringency levels and then assembling each data set with Velvet. A simple approach was devised to determine which filter strin- gency was “best.” Venn diagrams identified the regions containing reads that were used in an assembly but were of a low-enough quality to be removed by a filter. By plotting base quality histograms of reads in this region, we judged whether a filter was too stringent or not stringent enough. Our best assembly had a genome size of 33.6 Mb, an N50 of 65.8 kb for a k-mer of 51, and a maximum contig length of 347 kb. Using GeneMark, 9,262 genes were predicted. TargetP and SignalP analyses showed that among the 1,213 genes with secreted products, 986 had motifs for signal peptides and 227 had motifs for signal anchors. -

A New Species of Antrodia (Basidiomycota, Polypores) from China
Mycosphere 8(7): 878–885 (2017) www.mycosphere.org ISSN 2077 7019 Article Doi 10.5943/mycosphere/8/7/4 Copyright © Guizhou Academy of Agricultural Sciences A new species of Antrodia (Basidiomycota, Polypores) from China Chen YY, Wu F* Institute of Microbiology, Beijing Forestry University, Beijing 100083, China Chen YY, Wu F 2017 –A new species of Antrodia (Basidiomycota, Polypores) from China. Mycosphere 8(7), 878–885, Doi 10.5943/mycosphere/8/7/4 Abstract A new species, Antrodia monomitica sp. nov., is described and illustrated from China based on morphological characters and molecular evidence. It is characterized by producing annual, fragile and nodulose basidiomata, a monomitic hyphal system with clamp connections on generative hyphae, hyaline, thin-walled and fusiform to mango-shaped basidiospores (6–7.5 × 2.3– 3 µm), and causing a typical brown rot. In phylogenetic analysis inferred from ITS and nLSU rDNA sequences, the new species forms a distinct lineage in the Antrodia s. l., and has a close relationship with A. oleracea. Key words – Fomitopsidaceae – phylogenetic analysis – taxonomy – wood-decaying fungi Introduction Antrodia P. Karst., typified with Polyporus serpens Fr. (=Antrodia albida (Fr.) Donk (Donk 1960, Ryvarden 1991), is characterized by a resupinate to effused-reflexed growth habit, white or pale colour of the context, a dimitic hyphal system with clamp connections on generative hyphae, hyaline, thin-walled, cylindrical to very narrow ellipsoid basidiospores which are negative in Melzer’s reagent and Cotton Blue, and causing a brown rot (Ryvarden & Melo 2014). Antrodia is a highly heterogeneous genus which is closely related to Fomitopsis P. -

Three Species of Wood-Decaying Fungi in <I>Polyporales</I> New to China
MYCOTAXON ISSN (print) 0093-4666 (online) 2154-8889 Mycotaxon, Ltd. ©2017 January–March 2017—Volume 132, pp. 29–42 http://dx.doi.org/10.5248/132.29 Three species of wood-decaying fungi in Polyporales new to China Chang-lin Zhaoa, Shi-liang Liua, Guang-juan Ren, Xiao-hong Ji & Shuanghui He* Institute of Microbiology, Beijing Forestry University, No. 35 Qinghuadong Road, Haidian District, Beijing 100083, P.R. China * Correspondence to: [email protected] Abstract—Three wood-decaying fungi, Ceriporiopsis lagerheimii, Sebipora aquosa, and Tyromyces xuchilensis, are newly recorded in China. The identifications were based on morphological and molecular evidence. The phylogenetic tree inferred from ITS+nLSU sequences of 49 species of Polyporales nests C. lagerheimii within the phlebioid clade, S. aquosa within the gelatoporia clade, and T. xuchilensis within the residual polyporoid clade. The three species are described and illustrated based on Chinese material. Key words—Basidiomycota, polypore, taxonomy, white rot fungus Introduction Wood-decaying fungi play a key role in recycling nutrients of forest ecosystems by decomposing cellulose, hemicellulose, and lignin of the plant cell walls (Floudas et al. 2015). Polyporales, a large order in Basidiomycota, includes many important genera of wood-decaying fungi. Recent molecular studies employing multi-gene datasets have helped to provide a phylogenetic overview of Polyporales, in which thirty-four valid families are now recognized (Binder et al. 2013). The diversity of wood-decaying fungi is very high in China because of the large landscape ranging from boreal to tropical zones. More than 1200 species of wood-decaying fungi have been found in China (Dai 2011, 2012), and some a Chang-lin Zhao and Shi-liang Liu contributed equally to this work and share first-author status 30 .. -

Transcriptome Analysis of the Brown Rot Fungus Gloeophyllum Trabeum During Lignocellulose Degradation
University of Massachusetts Amherst ScholarWorks@UMass Amherst Microbiology Department Faculty Publication Series Microbiology 2020 Transcriptome analysis of the brown rot fungus Gloeophyllum trabeum during lignocellulose degradation Kiwamu Umezawa Mai Niikura Yuka Kojima Barry Goodell Makoto Yoshida Follow this and additional works at: https://scholarworks.umass.edu/micro_faculty_pubs PLOS ONE RESEARCH ARTICLE Transcriptome analysis of the brown rot fungus Gloeophyllum trabeum during lignocellulose degradation 1,2 1 1 3 1 Kiwamu UmezawaID *, Mai Niikura , Yuka Kojima , Barry Goodell , Makoto Yoshida 1 Department of Environmental and Natural Resource Science, Tokyo University of Agriculture and Technology, Tokyo, Japan, 2 Department of Applied Biological Chemistry, Kindai University, Nara, Japan, 3 Department of Microbiology, University of Massachusetts Amherst, Amherst, Massachusetts, United States of America a1111111111 a1111111111 * [email protected] a1111111111 a1111111111 a1111111111 Abstract Brown rot fungi have great potential in biorefinery wood conversion systems because they are the primary wood decomposers in coniferous forests and have an efficient lignocellulose OPEN ACCESS degrading system. Their initial wood degradation mechanism is thought to consist of an oxi- Citation: Umezawa K, Niikura M, Kojima Y, Goodell dative radical-based system that acts sequentially with an enzymatic saccharification sys- B, Yoshida M (2020) Transcriptome analysis of the tem, but the complete molecular mechanism of this system has not yet been elucidated. brown rot fungus Gloeophyllum trabeum during Some studies have shown that wood degradation mechanisms of brown rot fungi have lignocellulose degradation. PLoS ONE 15(12): diversity in their substrate selectivity. Gloeophyllum trabeum, one of the most studied brown e0243984. https://doi.org/10.1371/journal. pone.0243984 rot species, has broad substrate selectivity and even can degrade some grasses. -

A Phylogenetic Overview of the Antrodia Clade (Basidiomycota, Polyporales)
Mycologia, 105(6), 2013, pp. 1391–1411. DOI: 10.3852/13-051 # 2013 by The Mycological Society of America, Lawrence, KS 66044-8897 A phylogenetic overview of the antrodia clade (Basidiomycota, Polyporales) Beatriz Ortiz-Santana1 phylogenetic studies also have recognized the genera Daniel L. Lindner Amylocystis, Dacryobolus, Melanoporia, Pycnoporellus, US Forest Service, Northern Research Station, Center for Sarcoporia and Wolfiporia as part of the antrodia clade Forest Mycology Research, One Gifford Pinchot Drive, (SY Kim and Jung 2000, 2001; Binder and Hibbett Madison, Wisconsin 53726 2002; Hibbett and Binder 2002; SY Kim et al. 2003; Otto Miettinen Binder et al. 2005), while the genera Antrodia, Botanical Museum, University of Helsinki, PO Box 7, Daedalea, Fomitopsis, Laetiporus and Sparassis have 00014, Helsinki, Finland received attention in regard to species delimitation (SY Kim et al. 2001, 2003; KM Kim et al. 2005, 2007; Alfredo Justo Desjardin et al. 2004; Wang et al. 2004; Wu et al. 2004; David S. Hibbett Dai et al. 2006; Blanco-Dios et al. 2006; Chiu 2007; Clark University, Biology Department, 950 Main Street, Worcester, Massachusetts 01610 Lindner and Banik 2008; Yu et al. 2010; Banik et al. 2010, 2012; Garcia-Sandoval et al. 2011; Lindner et al. 2011; Rajchenberg et al. 2011; Zhou and Wei 2012; Abstract: Phylogenetic relationships among mem- Bernicchia et al. 2012; Spirin et al. 2012, 2013). These bers of the antrodia clade were investigated with studies also established that some of the genera are molecular data from two nuclear ribosomal DNA not monophyletic and several modifications have regions, LSU and ITS. A total of 123 species been proposed: the segregation of Antrodia s.l. -

Justo Et Al 2010 Pluteaceae.Pdf
ARTICLE IN PRESS fungal biology xxx (2010) 1e20 journal homepage: www.elsevier.com/locate/funbio Phylogeny of the Pluteaceae (Agaricales, Basidiomycota): taxonomy and character evolution Alfredo JUSTOa,*,1, Alfredo VIZZINIb,1, Andrew M. MINNISc, Nelson MENOLLI Jr.d,e, Marina CAPELARId, Olivia RODRıGUEZf, Ekaterina MALYSHEVAg, Marco CONTUh, Stefano GHIGNONEi, David S. HIBBETTa aBiology Department, Clark University, 950 Main St., Worcester, MA 01610, USA bDipartimento di Biologia Vegetale, Universita di Torino, Viale Mattioli 25, I-10125 Torino, Italy cSystematic Mycology & Microbiology Laboratory, USDA-ARS, B011A, 10300 Baltimore Ave., Beltsville, MD 20705, USA dNucleo de Pesquisa em Micologia, Instituto de Botanica,^ Caixa Postal 3005, Sao~ Paulo, SP 010631 970, Brazil eInstituto Federal de Educac¸ao,~ Ciencia^ e Tecnologia de Sao~ Paulo, Rua Pedro Vicente 625, Sao~ Paulo, SP 01109 010, Brazil fDepartamento de Botanica y Zoologıa, Universidad de Guadalajara, Apartado Postal 1-139, Zapopan, Jal. 45101, Mexico gKomarov Botanical Institute, 2 Popov St., St. Petersburg RUS-197376, Russia hVia Marmilla 12, I-07026 Olbia (OT), Italy iInstituto per la Protezione delle Piante, CNR Sezione di Torino, Viale Mattioli 25, I-10125 Torino, Italy article info abstract Article history: The phylogeny of the genera traditionally classified in the family Pluteaceae (Agaricales, Received 17 June 2010 Basidiomycota) was investigated using molecular data from nuclear ribosomal genes Received in revised form (nSSU, ITS, nLSU) and consequences for taxonomy and character evolution were evaluated. 16 September 2010 The genus Volvariella is polyphyletic, as most of its representatives fall outside the Pluteoid Accepted 26 September 2010 clade and shows affinities to some hygrophoroid genera (Camarophyllus, Cantharocybe). Corresponding Editor: Volvariella gloiocephala and allies are placed in a different clade, which represents the sister Joseph W.