Short-Read Sequencing for Genomic Analysis of the Brown Rot Fungus Fibroporia Radiculosa
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
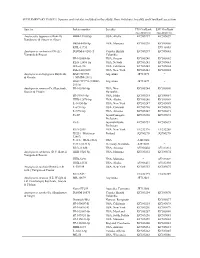
Suppl Table I
SUPPLEMENTARY TABLE I. Species and isolates included in the study, their vouchers, locality and GenBank accession Species Isolate number Locality ITS GenBank LSU GenBank accession no. accession no. Amylocystis lapponica (Romell) HHB-13400-Sp USA, Alaska KC585237 KC585059 Bondartsev & Singer ex Singer OKM-4418-Sp USA, Montana KC585238 KC585060 KHL-11755 − EU118603 Amyloporia carbonica (Overh.) DAOM-F-8281-T Canada, British KC585239 KC585061 Vampola & Pouzar Columbia FP-105585-Sp USA, Oregon KC585240 KC585062 RLG-12496-Sp USA, Nevada KC585241 KC585063 Wilcox-96 USA, California KC585242 KC585064 Zabel-40-GLN USA, New York KC585243 KC585065 Amyloporia nothofaginea Rajchenb. BAFC519794 Argentina JF713078 − & Gorjón (=MMBP-2011) BAFC519796 (=MBP- Argentina JF713079 − 2011a) Amyloporia sinuosa (Fr.) Rajchenb., FP-105386-Sp USA, New KC585244 KC585066 Gorjón & Pildain Hampshire FP-94464-Sp USA, Idaho KC585245 KC585067 HHB-12878-Sp USA, Alaska KC585246 KC585068 L-14130-Sp USA, New York KC585247 KC585069 L-6192-Sp USA, Colorado KC585248 KC585070 L-9792-Sp USA, Arizona KC585249 KC585071 Pa-3C JapanYamagata KC585250 KC585072 Prefecture Pa-3e JapanShizuoka KC585251 KC585073 Prefecture RLG-2538 USA, New York EU232196 EU232288 X725 (=Miettinen- Finland JQ700270 JQ700270 12407) P-115 (=RLG-2538) USA AJ416068 − P-211 (G-214) Germany, Karlsrube AJ345011 − RLG-1182R USA, Arizona AY966450 AY333831 Amyloporia sitchensis (D.V. Baxter) HHB-5320-Sp USA, Montana KC585252 KC585074 Vampola & Pouzar HHB-5298 USA, Montana − AY333829 HHB-12513 USA, Alaska AY966451 AY333830 -

Why Mushrooms Have Evolved to Be So Promiscuous: Insights from Evolutionary and Ecological Patterns
fungal biology reviews 29 (2015) 167e178 journal homepage: www.elsevier.com/locate/fbr Review Why mushrooms have evolved to be so promiscuous: Insights from evolutionary and ecological patterns Timothy Y. JAMES* Department of Ecology and Evolutionary Biology, University of Michigan, Ann Arbor, MI 48109, USA article info abstract Article history: Agaricomycetes, the mushrooms, are considered to have a promiscuous mating system, Received 27 May 2015 because most populations have a large number of mating types. This diversity of mating Received in revised form types ensures a high outcrossing efficiency, the probability of encountering a compatible 17 October 2015 mate when mating at random, because nearly every homokaryotic genotype is compatible Accepted 23 October 2015 with every other. Here I summarize the data from mating type surveys and genetic analysis of mating type loci and ask what evolutionary and ecological factors have promoted pro- Keywords: miscuity. Outcrossing efficiency is equally high in both bipolar and tetrapolar species Genomic conflict with a median value of 0.967 in Agaricomycetes. The sessile nature of the homokaryotic Homeodomain mycelium coupled with frequent long distance dispersal could account for selection favor- Outbreeding potential ing a high outcrossing efficiency as opportunities for choosing mates may be minimal. Pheromone receptor Consistent with a role of mating type in mediating cytoplasmic-nuclear genomic conflict, Agaricomycetes have evolved away from a haploid yeast phase towards hyphal fusions that display reciprocal nuclear migration after mating rather than cytoplasmic fusion. Importantly, the evolution of this mating behavior is precisely timed with the onset of diversification of mating type alleles at the pheromone/receptor mating type loci that are known to control reciprocal nuclear migration during mating. -

A New Species of Antrodia (Basidiomycota, Polypores) from China
Mycosphere 8(7): 878–885 (2017) www.mycosphere.org ISSN 2077 7019 Article Doi 10.5943/mycosphere/8/7/4 Copyright © Guizhou Academy of Agricultural Sciences A new species of Antrodia (Basidiomycota, Polypores) from China Chen YY, Wu F* Institute of Microbiology, Beijing Forestry University, Beijing 100083, China Chen YY, Wu F 2017 –A new species of Antrodia (Basidiomycota, Polypores) from China. Mycosphere 8(7), 878–885, Doi 10.5943/mycosphere/8/7/4 Abstract A new species, Antrodia monomitica sp. nov., is described and illustrated from China based on morphological characters and molecular evidence. It is characterized by producing annual, fragile and nodulose basidiomata, a monomitic hyphal system with clamp connections on generative hyphae, hyaline, thin-walled and fusiform to mango-shaped basidiospores (6–7.5 × 2.3– 3 µm), and causing a typical brown rot. In phylogenetic analysis inferred from ITS and nLSU rDNA sequences, the new species forms a distinct lineage in the Antrodia s. l., and has a close relationship with A. oleracea. Key words – Fomitopsidaceae – phylogenetic analysis – taxonomy – wood-decaying fungi Introduction Antrodia P. Karst., typified with Polyporus serpens Fr. (=Antrodia albida (Fr.) Donk (Donk 1960, Ryvarden 1991), is characterized by a resupinate to effused-reflexed growth habit, white or pale colour of the context, a dimitic hyphal system with clamp connections on generative hyphae, hyaline, thin-walled, cylindrical to very narrow ellipsoid basidiospores which are negative in Melzer’s reagent and Cotton Blue, and causing a brown rot (Ryvarden & Melo 2014). Antrodia is a highly heterogeneous genus which is closely related to Fomitopsis P. -

A Phylogenetic Overview of the Antrodia Clade (Basidiomycota, Polyporales)
Mycologia, 105(6), 2013, pp. 1391–1411. DOI: 10.3852/13-051 # 2013 by The Mycological Society of America, Lawrence, KS 66044-8897 A phylogenetic overview of the antrodia clade (Basidiomycota, Polyporales) Beatriz Ortiz-Santana1 phylogenetic studies also have recognized the genera Daniel L. Lindner Amylocystis, Dacryobolus, Melanoporia, Pycnoporellus, US Forest Service, Northern Research Station, Center for Sarcoporia and Wolfiporia as part of the antrodia clade Forest Mycology Research, One Gifford Pinchot Drive, (SY Kim and Jung 2000, 2001; Binder and Hibbett Madison, Wisconsin 53726 2002; Hibbett and Binder 2002; SY Kim et al. 2003; Otto Miettinen Binder et al. 2005), while the genera Antrodia, Botanical Museum, University of Helsinki, PO Box 7, Daedalea, Fomitopsis, Laetiporus and Sparassis have 00014, Helsinki, Finland received attention in regard to species delimitation (SY Kim et al. 2001, 2003; KM Kim et al. 2005, 2007; Alfredo Justo Desjardin et al. 2004; Wang et al. 2004; Wu et al. 2004; David S. Hibbett Dai et al. 2006; Blanco-Dios et al. 2006; Chiu 2007; Clark University, Biology Department, 950 Main Street, Worcester, Massachusetts 01610 Lindner and Banik 2008; Yu et al. 2010; Banik et al. 2010, 2012; Garcia-Sandoval et al. 2011; Lindner et al. 2011; Rajchenberg et al. 2011; Zhou and Wei 2012; Abstract: Phylogenetic relationships among mem- Bernicchia et al. 2012; Spirin et al. 2012, 2013). These bers of the antrodia clade were investigated with studies also established that some of the genera are molecular data from two nuclear ribosomal DNA not monophyletic and several modifications have regions, LSU and ITS. A total of 123 species been proposed: the segregation of Antrodia s.l. -

ABSTRACT BOOK Listed Alphabetically by Last Name Of
ABSTRACT BOOK Listed alphabetically by last name of presenting author AOS 2019 Meeting 24-28 June 2019 ORAL PRESENTATIONS Variability in the Use of Acoustic Space Between propensity, renesting intervals, and renest reproductive Two Tropical Forest Bird Communities success of Piping Plovers (Charadrius melodus) by fol- lowing 1,922 nests and 1,785 unique breeding adults Patrick J Hart, Kristina L Paxton, Grace Tredinnick from 2014 2016 in North and South Dakota, USA. The apparent renesting rate was 20%. Renesting propen- When acoustic signals sent from individuals overlap sity declined if reproductive attempts failed during the in frequency or time, acoustic interference and signal brood-rearing stage, nests were depredated, reproduc- masking occurs, which may reduce the receiver’s abil- tive failure occurred later in the breeding season, or ity to discriminate information from the signal. Under individuals had previously renested that year. Addi- the acoustic niche hypothesis (ANH), acoustic space is tionally, plovers were less likely to renest on reservoirs a resource that organisms may compete for, and sig- compared to other habitats. Renesting intervals de- naling behavior has evolved to minimize overlap with clined when individuals had not already renested, were heterospecific calling individuals. Because tropical after second-year adults without prior breeding experi- wet forests have such high bird species diversity and ence, and moved short distances between nest attempts. abundance, and thus high potential for competition for Renesting intervals also decreased if the attempt failed acoustic niche space, they are good places to examine later in the season. Lastly, overall reproductive success the way acoustic space is partitioned. -

Evidence of Subterranean Termite Feeding Deterrent Produced by Brown Rot Fungus Fibroporia Radiculosa (Peck) Parmasto 1968 (Polyporales, Fomitopsidaceae)
insects Article Evidence of Subterranean Termite Feeding Deterrent Produced by Brown Rot Fungus Fibroporia radiculosa (Peck) Parmasto 1968 (Polyporales, Fomitopsidaceae) Nadia Nuraniya Kamaluddin 1, Akiko Nakagawa-Izumi 1,*, Shota Nishizawa 1, Ayuko Fukunaga 1, Shuichi Doi 1, Tsuyoshi Yoshimura 2 and Sakae Horisawa 3 1 Graduate School of Life and Environmental Sciences, University of Tsukuba, Tsukuba 305-0872, Japan; [email protected](N.N.K.); [email protected], (S.N.); [email protected] (A.F.); [email protected] (S.D.) 2 Research Institute for Sustainable Humanosphere, Kyoto University, Uji, Kyoto 611-0011, Japan; [email protected] 3 School of Environmental Science and Engineering, Kochi University of Technology, Kami, Kochi 782-8502, Japan; [email protected] * Correspondence: [email protected]; Tel.: +81-29-853-4578 Academic Editor: Brian T. Forschler Received: 11 April 2016; Accepted: 9 August 2016; Published: 18 August 2016 Abstract: We found that decayed wood stakes with no termite damage collected from a termite-infested field exhibited a deterrent effect against the termite Reticulitermes speratus, Kolbe, 1885. The effect was observed to be lost or reduced by drying. After identification, it was found that the decayed stakes were infected by brown rot fungus Fibroporia radiculosa (Peck) Parmasto, 1968. In a no-choice feeding test, wood blocks decayed by this fungus under laboratory condition deterred R. speratus feeding and n-hexane extract from the decayed stake and blocks induced termite mortality. These data provided an insight into the interaction between wood-rot fungi and wood-feeding termites. -

Polypore Diversity in North America with an Annotated Checklist
Mycol Progress (2016) 15:771–790 DOI 10.1007/s11557-016-1207-7 ORIGINAL ARTICLE Polypore diversity in North America with an annotated checklist Li-Wei Zhou1 & Karen K. Nakasone2 & Harold H. Burdsall Jr.2 & James Ginns3 & Josef Vlasák4 & Otto Miettinen5 & Viacheslav Spirin5 & Tuomo Niemelä 5 & Hai-Sheng Yuan1 & Shuang-Hui He6 & Bao-Kai Cui6 & Jia-Hui Xing6 & Yu-Cheng Dai6 Received: 20 May 2016 /Accepted: 9 June 2016 /Published online: 30 June 2016 # German Mycological Society and Springer-Verlag Berlin Heidelberg 2016 Abstract Profound changes to the taxonomy and classifica- 11 orders, while six other species from three genera have tion of polypores have occurred since the advent of molecular uncertain taxonomic position at the order level. Three orders, phylogenetics in the 1990s. The last major monograph of viz. Polyporales, Hymenochaetales and Russulales, accom- North American polypores was published by Gilbertson and modate most of polypore species (93.7 %) and genera Ryvarden in 1986–1987. In the intervening 30 years, new (88.8 %). We hope that this updated checklist will inspire species, new combinations, and new records of polypores future studies in the polypore mycota of North America and were reported from North America. As a result, an updated contribute to the diversity and systematics of polypores checklist of North American polypores is needed to reflect the worldwide. polypore diversity in there. We recognize 492 species of polypores from 146 genera in North America. Of these, 232 Keywords Basidiomycota . Phylogeny . Taxonomy . species are unchanged from Gilbertson and Ryvarden’smono- Wood-decaying fungus graph, and 175 species required name or authority changes. -

Evaluation of Fungal Deterioration in Liquidambar Orientalis Mill
PEER-REVIEWED ARTICLE bioresources.com Evaluation of Fungal Deterioration in Liquidambar orientalis Mill. Heartwood by FT-IR and Light Microscopy Nural Yilgor,a* Dilek Dogu,b Roderquita Moore,c Evren Terzi,b and S. Nami Kartalb The chemical and morphological changes in heartwood specimens of Liquidambar orientalis Mill. caused by the white-rot fungus Trametes versicolor and the brown-rot fungi Tyromyces palustris and Gloeophyllum trabeum were studied by wet chemistry, FT-IR, GC-MS analyses, and photo-microscopy. According to GC-MS results, 26 extracts identified in the ethanol/toluene extraction and 17 in the ethanol extraction were found. Heartwood specimens of L. orientalis were highly susceptible to the fungi tested. While 1% NaOH solubility increased 35% in the specimen decayed by T. palustris, only an 8% increase was seen in the specimen exposed to T. versicolor when compared to the control specimen. Decayed wood by T. palustris showed a 5.5% increase in the Klason lignin content when compared to control specimens; however, the Klason lignin content decreased after a T. versicolor attack for 12 weeks. A T. versicolor attack in the cell walls was seen both from the lumina and from the cell corners, and the attack from the cell comers was mainly clear in ray parenchyma cells. An excessive destruction was detected in the wood structure attacked by T. palustris. The cell collapse was caused by a distortion in the plane of the wood cells. This extensive degradation was seen in all types of cell walls. Cracks in the cell walls were also detected in the specimens. -

Wood Research Indoor Fungal Destroyers of Wooden Materials – Their Identification in Present Review
WOOD RESEARCH 63 (2): 2018 203-214 INDOOR FUNGAL DESTROYERS OF WOODEN MATERIALS – THEIR IDENTIFICATION IN PRESENT REVIEW Ján Gáper Technical University in Zvolen, Faculty of Ecology and Environmental Sciences Department of Biology and Ecology Zvolen, Slovak Republic et University of Ostrava Faculty of Sciences, Department of Biology and Ecology Ostrava, Czech Republic Svetlana Gáperová, Terézia Gašparcová, Simona Kvasnová Matej Bel University, Faculty of Natural Sciences, Department of Biology and Ecology Banská Bystrica, Slovak Republic Peter Pristaš Pavol Jozef Šafárik University, Faculty of Natural Sciences Institute of Biology and Ecology Košice, Slovak Republic Kateřina Náplavová University of Ostrava, Faculty of Sciences, Department of Biology and Ecology Ostrava, Czech Republic et University of Minho Ceb-Centre of Biological Engineering, Campus De Gualtar Braga, Portugal (Received January 2018) ABSTRACT The wood-destroying fungi traditionally were separated from one another primarily on a basis of their sporocarp and/or strain morphology. Their diversity and simple macro- and 203 WOOD RESEARCH micromorphology of fungal structures have been major obstacles for more rapid progress in this regard. However, over the past two decades, there has been substantial progress in our understanding of genetic variability within traditionally recognized morphospecies. In this study we have overviewed genetic variation and phylogeography of macrofungi, which are important destroyers of wooden materials indoor of buildings. Several morphologically defined species of these fungal destroyers (Coniophora puteana, C. olivacea, C. arida, Serpula himantioides) have been shown to actually encompass several genetically isolated lineages (cryptic species). The protective efficacy against cryptic species within traditionally recognized morphospecies through laboratory tests (EN 113) and field trials (EN 252) might be sufficient to better prognosis of decay development in wooden materials for hazard assessment and for proper conservation and management plans. -

Phylogenetic Relationships of Antrodia Species and Related Taxa Based on Analyses of Nuclear Large Subunit Ribosomal DNA Sequences
Botanical Studies (2010) 51: 53-60. MicrobioLOGY Phylogenetic relationships of Antrodia species and related taxa based on analyses of nuclear large subunit ribosomal DNA sequences Zhi-HeYU1,Sheng-HuaWU2,*,Dong-MeiWANG3,andCheng-TauCHEN2 1College of Life Science, Yangtze University, Jingzhou 434025, Hubei, P.R. China 2Department of Botany, National Museum of Natural Science, Taichung 404, Taiwan 3Guangdong Provincial Key Laboratory of Microbial Culture Collection and Application, Guangdong Institute of Microbiology, Guangzhou 510070, P.R. China (Received March 24, 2008; Accepted July 10, 2009) ABSTRACT. ThisstudyaimedtoevaluaterelationshipsofAntrodia species and related taxa, including the taxonomic status of some Antrodiaspeciesthathavebeentreatedasseparategenera(Amyloporia,Fibroporia, andTaiwanofungus).AcomprehensivephylogeneticstudyofHomobasidiomycetespresentedbyBinderetal. in 2005, was consulted for sampling the taxa used for this analysis. The genera of the “residual” polyporoid cladeandphlebioidcladeoftheHomobasidiomyceteswerechosenasoutgroups,andthegenerabelonging totheAntrodiacladeandcorepolyporoidcladewereselectedasingroup.Phylogeneticanalysesofthisstudy werebasedonsequencedataderivedfromthenuclearlargesubunitribosomalDNA(nuc-LSUrDNA).The analytical methods of maximum parsimony (MP), maximum likelihood (ML), and Bayesian inference (BI) wereused.Resultsfromthesedifferentanalysesweregenerallyconsistent.Twomaincladeslackinghigh support were detected in the ingroup. Clade A consisted of taxa of the Antrodiaclade,includingalltwelve -

Characterization and Phylogenetic Analysis of the Complete Mitochondrial Genome of the Medicinal Fungus Laetiporus Sulphureus
www.nature.com/scientificreports OPEN Characterization and phylogenetic analysis of the complete mitochondrial genome of the Received: 29 December 2017 Accepted: 24 May 2018 medicinal fungus Laetiporus Published: xx xx xxxx sulphureus Qiang Li1,3, Mei Yang2, Cheng Chen4, Chuan Xiong1, Xin Jin1, Zhigang Pu1,5 & Wenli Huang1,5 The medicinal fungus Laetiporus sulphureus is widely distributed worldwide. To screen for molecular markers potentially useful for phylogenetic analyses of this species and related species, the mitochondrial genome of L. sulphureus was sequenced and assembled. The complete circular mitochondrial genome was 101,111 bp long, and contained 38 protein-coding genes (PCGs), 2 rRNA genes, and 25 tRNA genes. Our BLAST search aligned about 6.1 kb between the mitochondrial and nuclear genomes of L. sulphureus, indicative of possible gene transfer events. Both the GC and AT skews in the L. sulphureus mitogenome were negative, in contrast to the other seven Polyporales species tested. Of the 15 PCGs conserved across the seven species of Polyporales, the lengths of 11 were unique in the L. sulphureus mitogenome. The Ka/Ks of these 15 PCGs were all less than 1, indicating that PCGs were subject to purifying selection. Our phylogenetic analysis showed that three single genes (cox1, cob, and rnl) were potentially useful as molecular markers. This study is the frst publication of a mitochondrial genome in the family Laetiporaceae, and will facilitate the study of population genetics and evolution in L. sulphureus and other species in this family. Te fruiting body of Laetiporus sulphureus (Bull.) Murill, 1904 (Basidiomycota: Polyporales), with its striking citrus-yellow to pale orange color, is considered a cosmopolitan species, distributed from the boreal to tropical climactic zones1. -

Phylogenetic Position and Taxonomy of Kusaghiporia Usambarensis Gen
Mycology An International Journal on Fungal Biology ISSN: 2150-1203 (Print) 2150-1211 (Online) Journal homepage: http://www.tandfonline.com/loi/tmyc20 Phylogenetic position and taxonomy of Kusaghiporia usambarensis gen. et sp. nov. (Polyporales) Juma Mahmud Hussein, Donatha Damian Tibuhwa & Sanja Tibell To cite this article: Juma Mahmud Hussein, Donatha Damian Tibuhwa & Sanja Tibell (2018): Phylogenetic position and taxonomy of Kusaghiporia usambarensis gen. et sp. nov. (Polyporales), Mycology, DOI: 10.1080/21501203.2018.1461142 To link to this article: https://doi.org/10.1080/21501203.2018.1461142 © 2018 The Author(s). Published by Informa UK Limited, trading as Taylor & Francis Group. Published online: 15 Apr 2018. Submit your article to this journal View related articles View Crossmark data Full Terms & Conditions of access and use can be found at http://www.tandfonline.com/action/journalInformation?journalCode=tmyc20 MYCOLOGY, 2018 https://doi.org/10.1080/21501203.2018.1461142 Phylogenetic position and taxonomy of Kusaghiporia usambarensis gen. et sp. nov. (Polyporales) Juma Mahmud Husseina,b, Donatha Damian Tibuhwab and Sanja Tibella aInstitute of Organismal Biology, Evolutionary Biology Centre, Uppsala University, Uppsala, Sweden; bDepartment of Molecular Biology and Biotechnology, College of Natural & Applied Sciences, University of Dar es Salaam, Dar es Salaam, Tanzania ABSTRACT ARTICLE HISTORY A large polyporoid mushroom from the West Usambara Mountains in North-eastern Tanzania Received 17 November 2017 produces dark brown, up to 60-cm large fruiting bodies that at maturity may weigh more than Accepted 2 April 2018 10 kg. It has a high rate of mycelial growth and regeneration and was found growing on both dry KEYWORDS and green leaves of shrubs; attached to the base of living trees, and it was also observed to Kusaghiporia; molecular degrade dead snakes and insects accidentally coming into contact with it.