Genetic Diversity and RNA-Seq Transcriptome Analysis of Tricholoma Matsutake from Sichuan Province, China
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

World Bank Document
CONFORMED COPY Public Disclosure Authorized LOAN NUMBER 7616-CN Loan Agreement Public Disclosure Authorized (Wenchuan Earthquake Recovery Project) between PEOPLE’S REPUBLIC OF CHINA Public Disclosure Authorized and INTERNATIONAL BANK FOR RECONSTRUCTION AND DEVELOPMENT Dated March 20, 2009 Public Disclosure Authorized LOAN AGREEMENT AGREEMENT dated March 20, 2009, between PEOPLE’S REPUBLIC OF CHINA (“Borrower”) and INTERNATIONAL BANK FOR RECONSTRUCTION AND DEVELOPMENT (“Bank”). The Borrower and the Bank hereby agree as follows: ARTICLE I – GENERAL CONDITIONS; DEFINITIONS 1.01. The General Conditions (as defined in the Appendix to this Agreement) constitute an integral part of this Agreement. 1.02. Unless the context requires otherwise, the capitalized terms used in the Loan Agreement have the meanings ascribed to them in the General Conditions or in the Appendix to this Agreement. ARTICLE II – LOAN 2.01. The Bank agrees to lend to the Borrower, on the terms and conditions set forth or referred to in this Agreement, an amount equal to seven hundred ten million Dollars ($710,000,000), as such amount may be converted from time to time through a Currency Conversion in accordance with the provisions of Section 2.07 of this Agreement (“Loan”), to assist in financing the project described in Schedule 1 to this Agreement (“Project”). 2.02. The Borrower may withdraw the proceeds of the Loan in accordance with Section IV of Schedule 2 to this Agreement. 2.03. The Front-end Fee payable by the Borrower shall be equal to one quarter of one percent (0.25%) of the Loan amount. The Borrower shall pay the Front-end Fee not later than sixty (60) days after the Effective Date. -
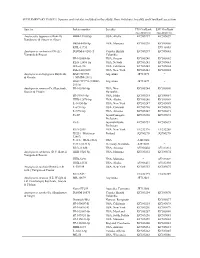
Suppl Table I
SUPPLEMENTARY TABLE I. Species and isolates included in the study, their vouchers, locality and GenBank accession Species Isolate number Locality ITS GenBank LSU GenBank accession no. accession no. Amylocystis lapponica (Romell) HHB-13400-Sp USA, Alaska KC585237 KC585059 Bondartsev & Singer ex Singer OKM-4418-Sp USA, Montana KC585238 KC585060 KHL-11755 − EU118603 Amyloporia carbonica (Overh.) DAOM-F-8281-T Canada, British KC585239 KC585061 Vampola & Pouzar Columbia FP-105585-Sp USA, Oregon KC585240 KC585062 RLG-12496-Sp USA, Nevada KC585241 KC585063 Wilcox-96 USA, California KC585242 KC585064 Zabel-40-GLN USA, New York KC585243 KC585065 Amyloporia nothofaginea Rajchenb. BAFC519794 Argentina JF713078 − & Gorjón (=MMBP-2011) BAFC519796 (=MBP- Argentina JF713079 − 2011a) Amyloporia sinuosa (Fr.) Rajchenb., FP-105386-Sp USA, New KC585244 KC585066 Gorjón & Pildain Hampshire FP-94464-Sp USA, Idaho KC585245 KC585067 HHB-12878-Sp USA, Alaska KC585246 KC585068 L-14130-Sp USA, New York KC585247 KC585069 L-6192-Sp USA, Colorado KC585248 KC585070 L-9792-Sp USA, Arizona KC585249 KC585071 Pa-3C JapanYamagata KC585250 KC585072 Prefecture Pa-3e JapanShizuoka KC585251 KC585073 Prefecture RLG-2538 USA, New York EU232196 EU232288 X725 (=Miettinen- Finland JQ700270 JQ700270 12407) P-115 (=RLG-2538) USA AJ416068 − P-211 (G-214) Germany, Karlsrube AJ345011 − RLG-1182R USA, Arizona AY966450 AY333831 Amyloporia sitchensis (D.V. Baxter) HHB-5320-Sp USA, Montana KC585252 KC585074 Vampola & Pouzar HHB-5298 USA, Montana − AY333829 HHB-12513 USA, Alaska AY966451 AY333830 -

Short-Read Sequencing for Genomic Analysis of the Brown Rot Fungus Fibroporia Radiculosa
Short-Read Sequencing for Genomic Analysis of the Brown Rot Fungus Fibroporia radiculosa Juliet D. Tang,a Andy D. Perkins,b Tad S. Sonstegard,c Steven G. Schroeder,c Shane C. Burgess,d and Susan V. Diehla Forest Products, Mississippi State University, Mississippi State, Mississippi, USAa; Computer Science and Engineering, Mississippi State University, Mississippi State, Mississippi, USAb; USDA ARS Bovine Functional Genomics Laboratory, Beltsville, Maryland, USAc; and Institute for Genomics, Biocomputing, and Biotechnology, Mississippi State University, Mississippi State, Mississippi, USAd The feasibility of short-read sequencing for genomic analysis was demonstrated for Fibroporia radiculosa, a copper-tolerant fungus that causes brown rot decay of wood. The effect of read quality on genomic assembly was assessed by filtering Illumina GAIIx reads from a single run of a paired-end library (75-nucleotide read length and 300-bp fragment size) at three different stringency levels and then assembling each data set with Velvet. A simple approach was devised to determine which filter strin- gency was “best.” Venn diagrams identified the regions containing reads that were used in an assembly but were of a low-enough quality to be removed by a filter. By plotting base quality histograms of reads in this region, we judged whether a filter was too stringent or not stringent enough. Our best assembly had a genome size of 33.6 Mb, an N50 of 65.8 kb for a k-mer of 51, and a maximum contig length of 347 kb. Using GeneMark, 9,262 genes were predicted. TargetP and SignalP analyses showed that among the 1,213 genes with secreted products, 986 had motifs for signal peptides and 227 had motifs for signal anchors. -

A New Species of Antrodia (Basidiomycota, Polypores) from China
Mycosphere 8(7): 878–885 (2017) www.mycosphere.org ISSN 2077 7019 Article Doi 10.5943/mycosphere/8/7/4 Copyright © Guizhou Academy of Agricultural Sciences A new species of Antrodia (Basidiomycota, Polypores) from China Chen YY, Wu F* Institute of Microbiology, Beijing Forestry University, Beijing 100083, China Chen YY, Wu F 2017 –A new species of Antrodia (Basidiomycota, Polypores) from China. Mycosphere 8(7), 878–885, Doi 10.5943/mycosphere/8/7/4 Abstract A new species, Antrodia monomitica sp. nov., is described and illustrated from China based on morphological characters and molecular evidence. It is characterized by producing annual, fragile and nodulose basidiomata, a monomitic hyphal system with clamp connections on generative hyphae, hyaline, thin-walled and fusiform to mango-shaped basidiospores (6–7.5 × 2.3– 3 µm), and causing a typical brown rot. In phylogenetic analysis inferred from ITS and nLSU rDNA sequences, the new species forms a distinct lineage in the Antrodia s. l., and has a close relationship with A. oleracea. Key words – Fomitopsidaceae – phylogenetic analysis – taxonomy – wood-decaying fungi Introduction Antrodia P. Karst., typified with Polyporus serpens Fr. (=Antrodia albida (Fr.) Donk (Donk 1960, Ryvarden 1991), is characterized by a resupinate to effused-reflexed growth habit, white or pale colour of the context, a dimitic hyphal system with clamp connections on generative hyphae, hyaline, thin-walled, cylindrical to very narrow ellipsoid basidiospores which are negative in Melzer’s reagent and Cotton Blue, and causing a brown rot (Ryvarden & Melo 2014). Antrodia is a highly heterogeneous genus which is closely related to Fomitopsis P. -

Study on the Ecotourism Development in Dazhou
Open Journal of Social Sciences, 2018, 6, 24-34 http://www.scirp.org/journal/jss ISSN Online: 2327-5960 ISSN Print: 2327-5952 Study on the Ecotourism Development in Dazhou Xiaomei Pu1, Lin Tian2, Zibiao Cheng3 1Research Center of Sichuan Old Revolutionary Areas Development, Sichuan University of Arts and Science, Dazhou, China 2School of Foreign Languages, Sichuan University of Arts and Science, Dazhou, China 3School of Finance and Economics Management, Sichuan University of Arts and Science, Dazhou, China How to cite this paper: Pu, X.M., Tian, L. Abstract and Cheng, Z.B. (2018) Study on the Eco- tourism Development in Dazhou. Open After comprehensive discussion of the origin of ecotourism, the concept of Journal of Social Sciences, 6, 24-34. ecotourism and the theoretical basis for ecotourism development, the paper https://doi.org/10.4236/jss.2018.65002 carried out the SWOT analysis on ecotourism development in Dazhou City, Received: April 8, 2018 and then proposed development strategies. The strategies were to: enhance Accepted: May 13, 2018 the ecological awareness of the entire people and create a good atmosphere for Published: May 16, 2018 ecotourism development; break the talent bottleneck of ecotourism develop- ment by adopting the policy of “combination boxing”; make scientific and Copyright © 2018 by authors and Scientific Research Publishing Inc. feasible master plan for Dazhou’s ecotourism development; develop quality This work is licensed under the Creative ecotourism products; innovate marketing strategies for ecotourism in Dazhou. Commons Attribution International License (CC BY 4.0). Keywords http://creativecommons.org/licenses/by/4.0/ Open Access Dazhou, Ecotourism, Development 1. -

Since the Reform and Opening Up1 1
Int. Statistical Inst.: Proc. 58th World Statistical Congress, 2011, Dublin (Session CPS020) p.6378 Research of Acceleration Urbanization Impacts on Resources and Environment in Sichuan Province Caimo,Teng National Bureau of Statistics of China, Survey Organizations of Sichuan No.31, the East Route, Qingjiang Road Chengdu, China, 610072 E-mail: [email protected] Since the reform and opening up, the rapid development of economic society and the rise ceaselessly of urbanization in Sichuan play an important role for material civilization and spiritual civilization, but also bring influence for resources and environment, this paper give an in-depth analysis about this. Ⅰ. The Main Characteristics of the Urbanization Development in Sichuan The reflection of urbanization in essence is from the industry cluster to population cluster., we tend to divided the process of urbanization into four stages, 1949-1978 is the first stage, 1978 – 1990 is the second stage, 1990 -2000 is the third stage, After the year of 2000 is the fourth stage. In view the particularities of the first phase, this paper researches mainly after three stages. 1. The level of the urbanization enhances unceasingly. With the reform and opening-up and the rapid development of social economy, the urbanization in Sichuan has significant achievements. The average annual growth of the level of urbanization is 0.8 percent in the twelve years of the second stage. The average annual growth in the third stage and the four stages is individually 0.5 and 1.3 percentage. The average annual growth of urbanization in the fourth stage is faster respectively 0.5 and 0.8 percent than the previous two stages which reflects obviously the rapid rise of the urbanization after the fourth stage in Sichuan. -

The Lichen Genus Hypogymnia in Southwest China Article
Mycosphere 5 (1): 27–76 (2014) ISSN 2077 7019 www.mycosphere.org Article Mycosphere Copyright © 2014 Online Edition Doi 10.5943/mycosphere/5/1/2 The lichen genus Hypogymnia in southwest China McCune B1 and Wang LS2 1 Department of Botany and Plant Pathology, Oregon State University, Corvallis, Oregon 97331-2902 U.S.A. 2 Key Laboratory of Biodiversity and Biogeography, Kunming Institute of Botany, Chinese Academy of Sciences, Heilongtan, Kunming 650204, China McCune B, Wang LS 2014 – The lichen genus Hypogymnia in southwest China. Mycosphere 5(1), 27–76, Doi 10.5943/mycosphere/5/1/2 Abstract A total of 36 species of Hypogymnia are known from southwestern China. This region is a center of biodiversity for the genus. Hypogymnia capitata, H. nitida, H. saxicola, H. pendula, and H. tenuispora are newly described species from Yunnan and Sichuan. Olivetoric acid is new as a major lichen substance in Hypogymnia, occurring only in H. capitata. A key and illustrations are given for the species known from this region, along with five species from adjoining regions that might be confused or have historically been misidentified in this region. Key words – Lecanorales – lichenized ascomycetes – Parmeliaceae – Shaanxi – Sichuan – Tibet – Yunnan – Xizang. Introduction The first major collections of Hypogymnia from southwestern China were by Handel- Mazzetti, from which Zahlbruckner (1930) reported six species now placed in Hypogymnia, and Harry Smith (1921-1934, published piecewise by other authors; Herner 1988). Since the last checklist of lichens in China (Wei 1991), which reported 16 species of Hypogymnia from the southwestern provinces, numerous species of Hypogymnia from southwestern China have been described or revised (Chen 1994, Wei & Bi 1998, McCune & Obermayer 2001, McCune et al. -

Sichuan Province
Directory of Important Bird Areas in China (Mainland): Key Sites for Conservation Editors SIMBA CHAN (Editor-in-chief) MIKE CROSBY , SAMSON SO, WANG DEZHI , FION CHEUNG and HUA FANGYUAN Principal compilers and data contributors Prof. Zhang Zhengwang (Beijing Normal University), Prof. Chang Jiachuan (Northeast Forestry University), the late Prof. Zhao Zhengjie (Forestry Institute of Jilin Province), Prof. Xing Lianlian (University of Nei Menggu), Prof. Ma Ming (Ecological and Geographical Institute, Chinese Academy of Sciences, Xinjiang), Prof. Lu Xin (Wuhan University), Prof. Liu Naifa (Lanzhou University), Prof. Yu Zhiwei (China West Normal University), Prof. Yang Lan (Kunming Institute for Zoology), Prof. Wang Qishan (Anhui University), Prof. Ding Changqing (Beijing Forestry University), Prof. Ding Ping (Zhejiang University), the late Prof. Gao Yuren (South China Institute for Endangered Animals), Prof. Zhou Fang (Guangxi University), Prof. Hu Hongxing (Wuhan University), Prof. Chen Shuihua (Zhejiang Natural History Museum), Tsering (Tibet University), Prof. Ma Zhijun (Fudan University), Prof. Guo Yumin (Capital Normal University), Dai Nianhua (Institute of Sciences, Jiangxi), Prof. Han Lianxian (Southwest Forestry University), Yang Xiaojun (Kunming Institute for Zoology), Prof. Wang Zijiang (Kunming Ornithological Association), Prof. Li Zhumei (Institute of Biology, Guizhou), Ma Chaohong (Management Office of Yellow River Wetland National Nature Reserve, Henan), Shen You (Chengdu Bird Watching Society), Wei Qian (Chengdu Bird Watching Society), Zhang Yu (Wild Bird Society of Jiangsu), Kang Hongli (Wild Bird Society of Shanghai). Information on Important Bird Areas in China was compiled with the support of the World Bank using consultant trust funds from the Government of Japan. Surveys of IBAs in western China were funded by Keidanren Nature Conservation Fund (Japan) and the Sekisui Chemical Co. -

Evidence of Subterranean Termite Feeding Deterrent Produced by Brown Rot Fungus Fibroporia Radiculosa (Peck) Parmasto 1968 (Polyporales, Fomitopsidaceae)
insects Article Evidence of Subterranean Termite Feeding Deterrent Produced by Brown Rot Fungus Fibroporia radiculosa (Peck) Parmasto 1968 (Polyporales, Fomitopsidaceae) Nadia Nuraniya Kamaluddin 1, Akiko Nakagawa-Izumi 1,*, Shota Nishizawa 1, Ayuko Fukunaga 1, Shuichi Doi 1, Tsuyoshi Yoshimura 2 and Sakae Horisawa 3 1 Graduate School of Life and Environmental Sciences, University of Tsukuba, Tsukuba 305-0872, Japan; [email protected](N.N.K.); [email protected], (S.N.); [email protected] (A.F.); [email protected] (S.D.) 2 Research Institute for Sustainable Humanosphere, Kyoto University, Uji, Kyoto 611-0011, Japan; [email protected] 3 School of Environmental Science and Engineering, Kochi University of Technology, Kami, Kochi 782-8502, Japan; [email protected] * Correspondence: [email protected]; Tel.: +81-29-853-4578 Academic Editor: Brian T. Forschler Received: 11 April 2016; Accepted: 9 August 2016; Published: 18 August 2016 Abstract: We found that decayed wood stakes with no termite damage collected from a termite-infested field exhibited a deterrent effect against the termite Reticulitermes speratus, Kolbe, 1885. The effect was observed to be lost or reduced by drying. After identification, it was found that the decayed stakes were infected by brown rot fungus Fibroporia radiculosa (Peck) Parmasto, 1968. In a no-choice feeding test, wood blocks decayed by this fungus under laboratory condition deterred R. speratus feeding and n-hexane extract from the decayed stake and blocks induced termite mortality. These data provided an insight into the interaction between wood-rot fungi and wood-feeding termites. -

The Transmission of Dazhou Folk Songs from Folk Philosophy in Dazhou City, Sichuan Province, China
The Transmission of Dazhou Folk Songs from Folk Philosophy in Dazhou City, Sichuan Province, China Chen Honglei A Thesis Submitted in Partial Fulfillment of Requirements for degree of Master of Music in Music February 2021 Copyright of Mahasarakham University การสืบทอดเพลงพ้ืนบา้ นตา้ โจว ตามวิถีปราชญ์ชาวบ้านในเมืองต้าโจว, เสฉวน, ประเทศจีน วิทยานิพนธ์ ของ Chen Honglei เสนอต่อมหาวทิ ยาลยั มหาสารคาม เพื่อเป็นส่วนหน่ึงของการศึกษาตามหลกั สูตร ปริญญาดุริยางคศาสตรมหาบัณฑิต สาขาวิชาดุริยางคศาสตรมหาบัณฑิต กุมภาพันธ์ 2564 ลิขสิทธ์ิเป็นของมหาวทิ ยาลยั มหาสารคาม The Transmission of Dazhou Folk Songs from Folk Philosophy in Dazhou City, Sichuan Province, China Chen Honglei A Thesis Submitted in Partial Fulfillment of Requirements for Master of Music (Music) February 2021 Copyright of Mahasarakham University The examining committee has unanimously approved this Thesis, submitted by Mr. Chen Honglei , as a partial fulfillment of the requirements for the Master of Music Music at Mahasarakham University Examining Committee Chairman (Asst. Prof. Khomkrit Karin , Ph.D.) Advisor (Asst. Prof. Peerapong Sensai , Ph.D.) Committee (Assoc. Prof. Phiphat Sornyai , Ph.D.) Committee (Assoc. Prof. Jatuporn Seemong , Ph.D.) Mahasarakham University has granted approval to accept this Thesis as a partial fulfillment of the requirements for the Master of Music Music (Asst. Prof. Khomkrit Karin , Ph.D.) (Assoc. Prof. Krit Chaimoon , Ph.D.) Dean of College of Music Dean of Graduate School D ABSTRACT TITLE The Transmission of Dazhou Folk Songs from Folk Philosophy in Dazhou City, Sichuan Province, China AUTHOR Chen Honglei ADVISORS Assistant Professor Peerapong Sensai , Ph.D. DEGREE Master of Music MAJOR Music UNIVERSITY Mahasarakham University YEAR 2021 ABSTRACT This research is "Folk Song Teaching in Parts of Dazhou City, Sichuan Province, China". The purpose of this article is to: 1 To study the transmission and collect information about Dazhou folk song from artists and procession of teaching Dazhou folk songs. -

SNOW LEOPARDS from the CAR, Qinghai
Qinghai 2017 Richard Webb SNOW LEOPARDS FROM THE CAR Qinghai: 27 February to 12 March 2017 Richard Webb CONTENTS Introduction Itinerary Logistics Mammal-watching sites 1. Guides 2. Flights Mammals 3. Visas 4. Accommodation Birds Qinghai Map INTRODUCTION I first saw Snow Leopard in Kazakhstan in 2003, well before Ladakh became the place to see the species. Plans to visit Ladakh have twice had to be aborted for health reasons and when it became apparent that Terry Townshend, and Jean-Michel Bompar (who had travelled with Sid Francis’s business partner, Roland) had both had success in southern Qinghai in 2016 I decided to give it a go with Sid Francis. 1 Qinghai 2017 Richard Webb It appeared a great move when we saw two leopards from the car within four hours of reaching the first site and although we only had one further sighting in the next nine days, we also heard leopards calling on at least two days and received what appeared to be reliable reports from yak herders of two leopards being seen in another of the valleys on a regular basis. It is unlikely that the area will ever rival Hemis in Ladakh in terms of its reliability for sightings given the number of well-trained leopard-spotters operating in that area. However the area appears to have potential with comfortable accommodation within 75 minutes’ drive of the sites and would be worth further investigation. I strongly recommend that anyone wanting to give it a try contacts Sid Francis to organise a trip at [email protected]. -

Mianyang Environmental Improvement Project
E1245 v 1 Sichuan Urban Development Project (SUDP) Financed by The World Bank Loan Public Disclosure Authorized Mianyang Environmental Improvement Project (Infrastructure and Access Improvement in Pioneer Park and Economic Development Zone) Public Disclosure Authorized Environmental Impact Assessment Report Public Disclosure Authorized (Draft for Review) Public Disclosure Authorized Sichuan Research Institute of Environmental Protection (SRIEP) September 2005 CONTENTS 1.0 INTRODUCTION …………………………………………… 1.1 Source and Necessity of the Proposed Project 1.2 Objectives, Principles and Methodology of the EIA 1.3 Policy, Legal and Administrative Framework 1.4 Standards for the EIA 1.5 Category of the EIA 1.6 Scope of the EIA 1.7 Factors of the EIA 1.8 Major Environmental Impacts and Protected Objects 1.9 Key Points of the EIA 1.10 Process and Procedure of the EIA 1.11 SRIEP and Staff of the EIA Core Team 2.0 PROJECT DESCRIPTION AND ANALYSeS …………………………………………………………….. 2.1 Project Description 2.2 Project Construction 2.3 Project Analysis 2.4 Conformity Analysis of Project and Local Development Plan 3.0 ENVIRONMENTAL SETTING ………………………….. (56) 3.1 Physical Environment 3.2 Socioeconomic Environment 3.3 Ecological Environment 3.4 Local Living Quality 3.5 Local Conditions of the Project Area 4.0 EXISTING ENVIRONMENTAL QUALITY ASSESSMENT ……………………………………………………………... (60) 4.1 Monitoring and Assessment of Existing Water Environment 4.2 Monitoring and Assessment of Existing Air Environment 4.3 Monitoring and Assessment of Existing Acoustic Environment 4.4