Are Universal Cox1 Gene Primers Too # #X201c
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

JMS 70 1 031-041 Eyh003 FINAL
PHYLOGENY AND HISTORICAL BIOGEOGRAPHY OF LIMPETS OF THE ORDER PATELLOGASTROPODA BASED ON MITOCHONDRIAL DNA SEQUENCES TOMOYUKI NAKANO AND TOMOWO OZAWA Department of Earth and Planetary Sciences, Nagoya University, Nagoya 464-8602,Japan (Received 29 March 2003; accepted 6June 2003) ABSTRACT Using new and previously published sequences of two mitochondrial genes (fragments of 12S and 16S ribosomal RNA; total 700 sites), we constructed a molecular phylogeny for 86 extant species, covering a major part of the order Patellogastropoda. There were 35 lottiid, one acmaeid, five nacellid and two patellid species from the western and northern Pacific; and 34 patellid, six nacellid and three lottiid species from the Atlantic, southern Africa, Antarctica and Australia. Emarginula foveolata fujitai (Fissurellidae) was used as the outgroup. In the resulting phylogenetic trees, the species fall into two major clades with high bootstrap support, designated here as (A) a clade of southern Tethyan origin consisting of superfamily Patelloidea and (B) a clade of tropical Tethyan origin consisting of the Acmaeoidea. Clades A and B were further divided into three and six subclades, respectively, which correspond with geographical distributions of species in the following genus or genera: (AÍ) north eastern Atlantic (Patella ); (A2) southern Africa and Australasia ( Scutellastra , Cymbula-and Helcion)', (A3) Antarctic, western Pacific, Australasia ( Nacella and Cellana); (BÍ) western to northwestern Pacific (.Patelloida); (B2) northern Pacific and northeastern Atlantic ( Lottia); (B3) northern Pacific (Lottia and Yayoiacmea); (B4) northwestern Pacific ( Nipponacmea); (B5) northern Pacific (Acmaea-’ânà Niveotectura) and (B6) northeastern Atlantic ( Tectura). Approximate divergence times were estimated using geo logical events and the fossil record to determine a reference date. -

The Limpets of Hong Kong with Descriptions of Seven New
60 DAVID DUDGEON Proceedings, First International Workshop Thompson, C.M. and Sparks, R.E. 1977b. Improbability of dispersal of adult Asiatic on the Malacofauna of clams, Corbicula manilensis via the intestinal tract of migratory waterfowl.American Hong Kong and Southern China, Midland Naturalist 98: 219-213. 23 March — 8 April 1977, Hong Kong Walford, P.R. 1946. A new graphic method of describing growth of animals.Biological Bulletin o f the Marine Biological Laboratory, Woods Hole 90: 141-147. Walne, P.R. 1972. The influence of current speed, body size and water temperature on the’ filtration rate o f five species o f bivalves.Journal o f the Marine Biological Asso THE LIMPETS OF HONG KONG WITH DESCRIPTIONS OF ciation of the United Kingdom 52: 345-374. SEVEN NEW SPECIES AND SUBSPECIES J. Christiaens 2 4 2 7 Justus Lipsiuslaan 26 B3 500, Hasselt, Belgium During the course of the malacological workshop the following stations were vistited: Wu Kwai Sha: a pebble beach, a rocky shore and a mangrove. Tolo Channel: Bluff Head (on the northern shore), Gruff Head (on the exposed southern shore) and Channel Rock, surrounded by coral, in the middle of the channel and reached by diving to a depth o f 10 m. Hong Kong Island: the exposed rocky shore at Wah Fu and the beach and bay at Stanley, the islands of Kat 0 Chau and Ping Chau, the last with a south-western shore exposed to heavy surf and sheltered to the north east. Subsequently two supplementary lots of limpets were received from Dr. -

Molecular Phylogeny and Historical Biogeography of Nacella (Patellogastropoda: Nacellidae) in the Southern Ocean
Molecular Phylogenetics and Evolution 56 (2010) 115–124 Contents lists available at ScienceDirect Molecular Phylogenetics and Evolution journal homepage: www.elsevier.com/locate/ympev Molecular phylogeny and historical biogeography of Nacella (Patellogastropoda: Nacellidae) in the Southern Ocean Claudio A. González-Wevar a, Tomoyuki Nakano b, Juan I. Cañete c, Elie Poulin a,* a Instituto de Ecología y Biodiversidad, Departamento de Ciencias Ecológicas, Facultad de Ciencias, Universidad de Chile, Las Palmeras # 3425, Ñuñoa, Santiago, Chile b Department of Geology and Paleontology, National Museum of Nature and Science, Tokyo, Japan c Departamento de Recursos Naturales, Universidad de Magallanes, Punta Arenas, Chile article info abstract Article history: The evolution and the historical biogeography of the Southern Ocean marine benthic fauna are closely Received 14 September 2009 related to major tectonic and climatic changes that occurred in this region during the last 55 million years Revised 29 January 2010 (Ma). Several families, genera and even species of marine organisms are shared between distant biogeo- Accepted 1 February 2010 graphic provinces in this region. This pattern of distribution in marine benthic invertebrates has been Available online 6 February 2010 commonly explained by vicariant speciation due to plate tectonics. However, recent molecular studies have provided new evidence for long-distance dispersion as a plausible explanation of biogeographical Keywords: patterns in the Southern Ocean. True limpets of the genus Nacella are currently distributed in different Antarctic Circumpolar Current biogeographic regions of the Southern Ocean such as Antarctica, Kerguelen Province, southern New Zea- Cellana Middle-Miocene climatic transition land Antipodean Province, North-Central Chile and South American Magellanic Province. -

Title STUDIES on the MOLLUSCAN FAECES (I) Author(S) Arakawa
Title STUDIES ON THE MOLLUSCAN FAECES (I) Author(s) Arakawa, Kohman Y. PUBLICATIONS OF THE SETO MARINE BIOLOGICAL Citation LABORATORY (1963), 11(2): 185-208 Issue Date 1963-12-31 URL http://hdl.handle.net/2433/175344 Right Type Departmental Bulletin Paper Textversion publisher Kyoto University STUDIES ON THE MOLLUSCAN FAECES (I)'l KoRMAN Y. ARAKAWA Miyajima Aquarium, Hiroshima, Japan With 7 Text-figures Since Lister (1678) revealed specific differences existing among some molluscan faecal pellets, several works on the same line have been published during last three decades by various authors, i.e. MooRE (1930, '31, '31a, '31b, '32, '33, '33a, '39), MANNING & KuMPF ('59), etc. in which observations are made almost ex clusively upon European and American species. But yet our knowledge about this subject seems to be far from complete. Thus the present work is planned to enrich the knowledge in this field and based mainly on Japanese species as many as possible. In my previous paper (ARAKAWA '62), I have already given a general account on the molluscan faeces at the present level of our knowledge in this field to gether with my unpublished data, and so in the first part of this serial work, I am going to describe and illustrate in detail the morphological characters of faecal pellets of molluscs collected in the Inland Sea of Seto and its neighbour ing areas. Before going further, I must express here my hearty thanks first to the late Dr. IsAo TAKI who educated me to carry out works in Malacology as one of his pupils, and then to Drs. -

Bacterial Diversity in the Intestine of Sea Cucumber Stichopus Japonicus
Bacterial diversity in the intestine of sea cucumber Stichopus japonicus Item Type article Authors Gao, M.L.; Hou, H.M.; Zhang, G.L.; Liu, Y.; Sun, L.M. Download date 29/09/2021 15:37:50 Link to Item http://hdl.handle.net/1834/37808 Iranian Journal of Fisheries Sciences 16(1)318-325 2017 Bacterial diversity in the intestine of sea cucumber Stichopus japonicus * Gao M.L.; Hou H.M. ; Zhang G.L.; Liu Y.; Sun L.M. Received: May 2015 Accepted: August 2016 Abstract The intestinal bacterial diversity of Stichopus japonicus was investigated using 16S ribosomal RNA gene (rDNA) clone library and Polymerase Chain Reaction/Denaturing Gradient Gel Electrophoresis (PCR-DGGE). The clone library yielded a total of 188 clones, and these were sequenced and classified into 106 operational taxonomic units (OTUs) with sequence similarity ranging from 88 to 100%. The coverage of the library was 77.4%, with approximately 88.7% of the sequences affiliated to Proteobacteria. Gammaproteobacteria and Vibrio sp. were the predominant groups in the intestine of S. japonicus. Some bacteria such as Legionella sp., Brachybacterium sp., Streptomyces sp., Propionigenium sp. and Psychrobacter sp were first identified in the intestine of sea cucumber. Keywords: Intestinal bacterial diversity, 16S rDNA, PCR-DGGE, Sequencing, Stichopus japonicus School of Food Science and Technology- Dalian Polytechnic University, Dalian 116034, P. R. China * Corresponding author's Email:[email protected] 319 Gao et al., Bacterial diversity in the intestine of sea cucumber Stichopus japonicas Introduction In this paper, 16S rDNA clone Sea cucumber Stichopus japonicus is library analysis approaches and one of the most important holothurian PCR-DGGE fingerprinting of the 16S species in coastal fisheries. -

Isolation and Characterization of a Novel Agar-Degrading Marine Bacterium, Gayadomonas Joobiniege Gen, Nov, Sp
J. Microbiol. Biotechnol. (2013), 23(11), 1509–1518 http://dx.doi.org/10.4014/jmb.1308.08007 Research Article jmb Isolation and Characterization of a Novel Agar-Degrading Marine Bacterium, Gayadomonas joobiniege gen, nov, sp. nov., from the Southern Sea, Korea Won-Jae Chi1, Jae-Seon Park1, Min-Jung Kwak2, Jihyun F. Kim3, Yong-Keun Chang4, and Soon-Kwang Hong1* 1Division of Biological Science and Bioinformatics, Myongji University, Yongin 449-728, Republic of Korea 2Biosystems and Bioengineering Program, University of Science and Technology, Daejeon 305-350, Republic of Korea 3Department of Systems Biology, Yonsei University, Seoul 120-749, Republic of Korea 4Department of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology, Daejeon 305-701, Republic of Korea Received: August 2, 2013 Revised: August 14, 2013 An agar-degrading bacterium, designated as strain G7T, was isolated from a coastal seawater Accepted: August 20, 2013 sample from Gaya Island (Gayado in Korean), Republic of Korea. The isolated strain G7T is gram-negative, rod shaped, aerobic, non-motile, and non-pigmented. A similarity search based on its 16S rRNA gene sequence revealed that it shares 95.5%, 90.6%, and 90.0% T First published online similarity with the 16S rRNA gene sequences of Catenovulum agarivorans YM01, Algicola August 22, 2013 sagamiensis, and Bowmanella pacifica W3-3AT, respectively. Phylogenetic analyses demonstrated T *Corresponding author that strain G7 formed a distinct monophyletic clade closely related to species of the family Phone: +82-31-330-6198; Alteromonadaceae in the Alteromonas-like Gammaproteobacteria. The G+C content of strain Fax: +82-31-335-8249; G7T was 41.12 mol%. -

DNA Barcoding Reveal Patterns of Species Diversity Among
www.nature.com/scientificreports OPEN DNA barcoding reveal patterns of species diversity among northwestern Pacific molluscs Received: 04 April 2016 Shao’e Sun, Qi Li, Lingfeng Kong, Hong Yu, Xiaodong Zheng, Ruihai Yu, Lina Dai, Yan Sun, Accepted: 25 August 2016 Jun Chen, Jun Liu, Lehai Ni, Yanwei Feng, Zhenzhen Yu, Shanmei Zou & Jiping Lin Published: 19 September 2016 This study represents the first comprehensive molecular assessment of northwestern Pacific molluscs. In total, 2801 DNA barcodes belonging to 569 species from China, Japan and Korea were analyzed. An overlap between intra- and interspecific genetic distances was present in 71 species. We tested the efficacy of this library by simulating a sequence-based specimen identification scenario using Best Match (BM), Best Close Match (BCM) and All Species Barcode (ASB) criteria with three threshold values. BM approach returned 89.15% true identifications (95.27% when excluding singletons). The highest success rate of congruent identifications was obtained with BCM at 0.053 threshold. The analysis of our barcode library together with public data resulted in 582 Barcode Index Numbers (BINs), 72.2% of which was found to be concordantly with morphology-based identifications. The discrepancies were divided in two groups: sequences from different species clustered in a single BIN and conspecific sequences divided in one more BINs. In Neighbour-Joining phenogram, 2,320 (83.0%) queries fromed 355 (62.4%) species-specific barcode clusters allowing their successful identification. 33 species showed paraphyletic and haplotype sharing. 62 cases are represented by deeply diverged lineages. This study suggest an increased species diversity in this region, highlighting taxonomic revision and conservation strategy for the cryptic complexes. -

Two New Species of Patelloida (Patellogastropoda: Lottiidae) from West Java, Indonesia
VENUS 66 (3-4): 105-111, 2008 Two New Species of Patelloida (Patellogastropoda: Lottiidae) from West Java, Indonesia Tomoyuki Nakano1 and Aswan2 1National Museum of Nature and Science, 3-23-1 Hyakunin-cho, Shinjuku-ku, Tokyo 169-0073, Japan; [email protected] 2Department of Geology, Faculty of Earth Science and Technology, Institut Teknologi Bandung (ITB), Bandung 40132, Indonesia Abstract: Nine species of patellogastropod limpets belonging to three families and four genera and two additional undescribed species are distributed in West Java, Indonesia. We formally name these two Patelloida species herein, based on shell and anatomical characters. Keywords: Patelloida garuda n. sp., Patelloida pseudopygmaea n. sp., Lottiidae, West Java Introduction Patellogastropod limpets are a diverse group of mollusks occurring throughout the world oceans from tropical to polar regions. The genus Patelloida reaches its highest diversity in the Australasian region (Ponder & Creese, 1980). Nine species are currently recognized in Indonesia, belonging to Cellana, Eoacmaea, Lottia, Patelloida and Scutellastra (Dharma, 1988, 2005; Nakano et al., 2005). According to Nakano et al. (2005), there are two additional yet unidentified new species in Java. Recent molecular work by Nakano & Ozawa (2007) revealed that these two species are genetically distinct from other related species, and they were temporarily named Patelloida ESU3 and Patelloida ESU4. The purpose of this paper is to describe formally these two new Patelloida species. Abbreviations: NSMT - National Museum of Nature and Science, Tokyo, Japan Materials and Methods We collected limpets in rocky shores between Pelabuan Ratu and Jakarta, West Java, Indonesia during October, 2004. The material used in this paper was collected in the Indian Ocean at Pelabuan Ratu (06˚59.3´S, 106˚32.4´E), Karang Hawu Beach (06˚57.3´S, 106˚27.5´E), Sedekan Beach (06˚52.9´S, 106˚05´E) and Carita Beach (6˚16.32´S, 105˚49.74´E) from calcareous substrata in the high intertidal to supratidal zones and on rocky shores in the intertidal zone. -

Ecological Roles of Arc Signal Transduction System Revealed by Evolutionary Genetics Analysis
Open Access Journal of Bacteriology and Mycology Research Article Ecological Roles of Arc Signal Transduction System Revealed by Evolutionary Genetics Analysis Yangyang Dong, Fen Wan, Jianhua Yin and Haichun Gao* Abstract Institute of Microbiology and College of Life Sciences, The Arc signal transduction system was studied in γ-proteobacteria to Zhejiang University, China determine evolutionary mechanisms and ecological pressures responsible for *Corresponding author: Haichun Gao, Institute the plasticity of this system. Phylogenic analysis of the arcA gene encoding the of Microbiology and College of Life Sciences, Zhejiang DNA-binding response regulator suggests that the gene has remained under University, China strong purifying selection throughout its history while the arcB gene encoding the sensor histidine kinase has undergone extensive modification in various Received: August 14, 2014; Accepted: November 06, lineages. The major observed modification occurs in the Alteromonadaceae 2014; Published: November 12, 2014 family, where the Hpt domain of ArcB has been uncoupled from the sensor component in several genera (Shewanella, Colwellia and Idiomarina). This phenomenon appears to be linked to the free-living lifestyles of the Alteromonads. In contrast to the prevailing view of the Arc system existing only in facultative anaerobes, several of the Alteromonad species which encode Arc systems are strict aerobes or aero-tolerant anaerobes, suggesting additional functions for the Arc system beyond facultative anaerobiosis. Molecular clock estimates using linearized phylogenetics trees for the arcA gene place the origins of the system at 600-1300 million years ago, a period roughly corresponding to the rise in atmospheric oxygen levels that occurred in the Precambrian Era. It is hypothesized that the Arc system originated as an early oxidative stress response mechanism in the γ-proteobacteria and was later co-opted for the facultative anaerobic lifestyle. -
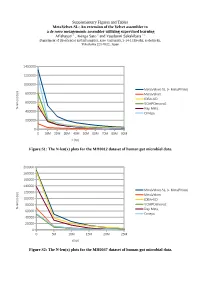
Supplementary Figures and Tables Metavelvet-SL
Supplementary Figures and Tables MetaVelvet-SL: An extension of the Velvet assembler to a de novo metagenomic assembler utilizing supervised learning Afiahayati 1 , Kengo Sato 1 and Yasubumi Sakakibara 1∗ Department of Biosciences and Informatics, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama 223-8522, Japan 1400000 1200000 1000000 MetaVelvet-SL (+ MetaPhlAn) ) p 800000 b MetaVelvet ( ) x ( IDBA-UD n 600000 e SOAPDenovo2 l - N Ray Meta 400000 Omega 200000 0 0 10M 20M 30M 40M 50M 60M 70M 80M 90M x (bp) Figure S1: The N-len(x) plots for the MH0012 dataset of human gut microbial data. 200000 180000 160000 140000 MetaVelvet-SL (+ MetaPhlAn) ) 120000 p b MetaVelvet ( ) 100000 x IDBA-UD ( n e l 80000 SOAPDenovo2 - N Ray Meta 60000 Omega 40000 20000 0 0 5M 10M 15M 20M 25M x(bp) Figure S2: The N-len(x) plots for the MH0047 dataset of human gut microbial data. 600000 500000 400000 MetaVelvet-SL (+ MetaPhlAn) ) p b MetaVelvet ( ) 300000 x IDBA-UD ( n e l SOAPDenovo2 - N 200000 Ray Meta Omega 100000 0 0 10M 20M 30M 40M 50M 60M 70M 80M 90M x (bp) Figure S3: The N-len(x) plots for the SRS017227 dataset of human gut microbial data. 800000 700000 600000 500000 MetaVelvet-SL (+ MetaPhlAn) ) p b MetaVelvet ( ) 400000 x IDBA-UD ( n e l SOAPDenovo2 - 300000 N Ray Meta 200000 Omega 100000 0 0 5M 10M 15M 20M 25M 30M 35M 40M 45M x (bp) Figure S4: The N-len(x) plots for the SRS018661 dataset of human gut microbial data. Formula to identify unique nodes in Velvet (Zerbino and Birney, 2008) ̄x2 ρ2− log2 2 F ( ̄x ,n,ρ )= +n 2 2 where ̄x = the coverage of node ρ = expected coverage of subgraph n = the length of node A node is “unique”, if its F > 5 . -

Supplementary 3
TROPICAL NATURAL HISTORY Department of Biology, Faculty of Science, Chulalongkorn University Editor: SOMSAK PANHA ([email protected]) Department of Biology, Faculty of Science, Chulalongkorn University, Bangkok 10330, THAILAND Consulting Editor: FRED NAGGS, The Natural History Museum, UK Associate Editors: PONGCHAI HARNYUTTANAKORN, Chulalongkorn University, THAILAND WICHASE KHONSUE, Chulalongkorn University, THAILAND KUMTHORN THIRAKHUPT, Chulalongkorn University, THAILAND Assistant Editors: NONTIVITCH TANDAVANIJ, Chulalongkorn University, THAILAND PIYOROS TONGKERD, Chulalongkorn University, THAILAND CHIRASAK SUTCHARIT, Chulalongkorn University, THAILAND Editorial Board TAKAHIRO ASAMI, Shinshu University, JAPAN DON L. MOLL, Southwest Missouri State University, USA VISUT BAIMAI, Mahidol University, THAILAND PHAIBUL NAIYANETR, Chulalongkorn University, BERNARD R. BAUM, Eastern Cereal and Oilseed Research THAILAND Centre, CANADA PETER K.L. NG, National University of Singapore, ARTHUR E. BOGAN, North Corolina State Museum of SINGAPORE Natural Sciences, USA BENJAMIN P. OLDROYD, The University of Sydney, THAWEESAKDI BOONKERD, Chulalongkorn University, AUSTRALIA THAILAND HIDETOSHI OTA, Museum of Human and Nature, University WARREN Y. BROCKELMAN, Mahidol University, of Hyogo, JAPAN THAILAND PETER C.H. PRITCHARD, Chelonian Research Institute, JOHN B. BURCH, University of Michigan, USA USA PRANOM CHANTARANOTHAI, Khon Kaen University, DANIEL ROGERS, University of Adelaide, AUSTRALIA THAILAND DAVID A. SIMPSON, Herbarium, Royal Botanic Gardens, -

Bacterial Taxa Based on Greengenes Database GS1A PS1B ABY1 OD1
A1: Bacterial taxa based on GreenGenes database GS1A PS1B ABY1_OD1 0.1682 0.024 Bacteria;ABY1_OD1;ABY1_OD1_unclassified 1 0 Bacteria;ABY1_OD1;FW129;FW129_unclassified 4 0 Bacteria;ABY1_OD1;FW129;KNA6-NB12;KNA6-NB12_unclassified 5 0 Bacteria;ABY1_OD1;FW129;KNA6-NB29;KNA6-NB29_unclassified 0 1 Acidobacteria 0.7907 4.509 Bacteria;Acidobacteria;Acidobacteria_unclassified 4 31 Bacteria;Acidobacteria;Acidobacteria-5;Acidobacteria-5_unclassified 0 1 Bacteria;Acidobacteria;BPC015;BPC015_unclassified 8 30 Bacteria;Acidobacteria;BPC102;BPC102_unclassified 9 43 Bacteria;Acidobacteria;Chloracidobacteria;Ellin6075;Ellin6075_unclassified 1 0 Bacteria;Acidobacteria;iii1-15;Acidobacteria-6;RB40;RB40_unclassified 0 5 Bacteria;Acidobacteria;iii1-15;iii1-15_unclassified 1 8 Bacteria;Acidobacteria;iii1-15;Riz6I;Unclassified 0 1 Bacteria;Acidobacteria;iii1-8;Unclassified 0 2 Bacteria;Acidobacteria;OS-K;OS-K_unclassified 18 17 Bacteria;Acidobacteria;RB25;RB25_unclassified 6 47 Bacteria;Acidobacteria;Solibacteres;Solibacteres_unclassified 0 1 Actinobacteria 2.1198 6.642 Bacteria;Actinobacteria;Acidimicrobidae;Acidimicrobidae_unclassified 10 70 Bacteria;Actinobacteria;Acidimicrobidae;CL500-29;ML316M-15;ML316M-15_unclassified 0 3 Bacteria;Actinobacteria;Acidimicrobidae;EB1017_group;Acidimicrobidae_bacterium_Ellin7143;Unclassified 6 1 Bacteria;Actinobacteria;Acidimicrobidae;koll13;JTB31;BD2-10;BD2-10_unclassified 1 5 Bacteria;Actinobacteria;Acidimicrobidae;koll13;JTB31;Unclassified 16 37 Bacteria;Actinobacteria;Acidimicrobidae;koll13;koll13_unclassified 81 25 Bacteria;Actinobacteria;Acidimicrobidae;Microthrixineae;Microthrixineae_unclassified